LC-MS/MS 法测定人血浆中沙丙蝶呤的浓度
2021-07-13王月玥林明弘
王月玥,林明弘,2*
1 徐州医科大学,徐州 221000;2 徐州立兴佳正医药科技有限公司,徐州 221009
沙丙蝶呤(Sapropterin),也称四氢生物蝶呤(Tetrahydrobiopterin,BH4),是一氧化氮合酶的辅助因子,是一种内源性物质,存在于高等动物每个细胞和组织中,参与多个生理和病理过程,在酶促反应中作为电子载体,起到还原剂的作用。BH4很容易被氧化成二氢生物蝶呤(Dihydrobiopterin,BH2)和生物蝶呤(Biopterin,B),BH2也容易被氧化成B;但只有BH4能够提供合适的辅助因子功能。BH4不足或催化BH4再生的还原酶缺陷,是导致苯丙氨酸羟化反应受阻、出现苯丙酮尿症的原因之一[1],目前已有相应的药物制剂应用于临床治疗[2]。盐酸沙丙蝶呤(sapropterin dihydrochloride)是美国FDA 批准的首个治疗由于BH4缺乏所导致苯丙酮尿症的特异性药物[3]。该药物疗效确切、服用方便、安全性较高[4];但关于此药的相关报道较少,因此有必要建立快速、简便、可靠的人体血药浓度测定方法,有利于开展BH4的药代动力学研究,为制剂开发和临床合理用药提供指导,保证用药安全。
沙丙蝶呤不稳定,很容易被氧化,这对血液样本的处理和分析造成较大的难度。近年来,国内外多采用选择性好、分析速度快的液相色谱串联质谱法(LC-MS/MS)来测定沙丙蝶呤的血药浓度。根据现有文献[5-8]报道,通过测定血浆中B 的浓度来间接确定BH4的浓度[7],在处理生物样本的过程中采取低温萃取或者通过衍生化等处理方法[5];但样本处理过程都比较繁琐,分析时间普遍较长,线性范围较小,不适用于大量临床或非临床样本的分析。
本研究采用较为简便的蛋白质沉淀法处理生物样本,在此过程中分批次加入不同浓度的抗氧化剂(抗坏血酸,VC)用于稳定BH4,避免发生氧化反应,从而能够直接测定人血浆中的BH4浓度,具有稳定性好、专属性强、灵敏度高、样本处理简单的特点。
1 材 料
1.1 药品与试剂
对照品:沙丙蝶呤(来源:Clearsynth Inspiring Research;批号:CS-WS-AAA-1982-02;纯度:99.44%);沙丙蝶呤-13C15N3(来源:Clearsynth Inspiring Research;批号:CS-SAT-426;纯度:91.18%(Purity),97.25%(Isotopic enrichment))。受试制剂:盐酸沙丙蝶呤片(盐酸沙丙蝶呤,100 mg,口服片剂,市售);参比制剂:Kuvan®(盐酸沙丙蝶呤,100 mg,口服片剂,自制)。
乙酸铵、甲醇、乙腈(制造商:J.T.Baker,级别:HPLC);抗坏血酸(VC,制造商:SIGMA-ALDRICH,级别:ACS);盐酸(制造商:Greagent,级别:AR);纯水由德国Sartorius 公司的纯水制造机制备。
人空白血浆来源:购买和采集于志愿者。
1.2 仪器
液相色谱串联质谱联用仪(美国AB SCIEX 公司):包括液相泵(型号:AC Pump)、脱气机(型号:AC Degasser)、自动进样器(型号:AC Autosampler)、柱温箱(型号:AC Column Oven)、样本架(型号:AC Rack Changer)、数据处理系统(Analyst 1.6.3(software))、三重四级杆质谱仪(型号:QTRAP®6500+);百万分之一天平(德国Sartorius 公司,型号:MSA6.6S-0CE-DM);千分之一天平(德国Sartorius 公司,型号:BSA623S);离心机(德国HERMLE公司,型号:Z513K);多试管涡旋混合仪(中国米欧仪器有限公司,型号:DMT-2500)。
2 方 法
2.1 色谱条件
色谱柱:Kinetex® EVO C18100A˚(4.6 mm×100 mm,5 μm);柱温:30 ℃;流动相:含0.1% 1 mol·L-1的乙酸铵水溶液∶乙腈=93∶7(v/v);自动进样器温度:6 ℃;流速:0.6 mL·min-1;进样量:15 μL;分析时间:3 min。
2.2 质谱条件
采用电喷雾离子源(ESI),正离子模式检测,扫描方式为多反应检测(MRM)。仪器参数设置为:气帘气压 力(CUR):138 kPa,碰撞气压 力(CAD):High,离子源喷射电压(IS):2800 V,温度(TEM):650 ℃,雾化气压力(GS1):586 kPa,辅助气压力(GS2):586 kPa,去簇电压(DP):20 eV,碰撞室入口电压(EP):10 eV,碰撞能量(CE):26 eV,碰撞室出口电压(CXP):10 eV。沙丙蝶呤和内标沙丙蝶呤-13C15N3的检测目标离子对分别为m/z 242.3→166.1和m/z 246.1→170.2。
2.3 溶液的配制
2.3.1 抗氧化剂溶液 称取VC 约12.5g,用含0.1mol·L-1盐酸的甲醇水溶液(甲醇∶水=50∶50,v/v)定容至250 mL,配制成5%的VC 溶液;称量VC 约50 g,用含0.1 mol·L-1盐酸的甲醇水溶液(甲醇∶水=1∶99,v/v)定容至250 mL,配制成20%的VC 溶液Ⅰ;称量VC 约50 g,用含0.02 mol·L-1盐酸的甲醇水溶液(甲醇∶水=1∶99,v/v)定容至250 mL,配制成20%的VC溶液Ⅱ。由于VC 遇光、热不稳定,故配制过程需在避光条件下进行,配制好的溶液于4 ℃冷藏柜避光保存。
2.3.2 沙丙蝶呤储备液 精密称取沙丙蝶呤对照品1.025 mg,用5%的VC 溶液溶解并稀释配制成浓度约为100 ng·μL-1的储备液-Ⅰ。再精密称取沙丙蝶呤对照品1.019 mg,用5%的VC 溶液溶解并稀释配制成浓度约为100 ng·μL-1的储备液-Ⅱ,配制过程在避光条件下进行,配制好的溶液于-20 ℃冰箱避光保存。
2.3.3 内标溶液 精密称取沙丙蝶呤-13C15N31.115mg,用5%的VC 溶液溶解并稀释配制成浓度约为50 ng·μL-1的储备液,用5%的VC 溶液稀释储备液,配制成0.2 ng·μL-1的内标标准溶液。配制过程在避光条件下进行,配制好的溶液于-20 ℃冰箱避光保存。
2.4 血浆样品处理
在96 深孔板中加入含药血浆样品100 μL,添加0.2 ng·μL-1内标标准溶液5 μL 和20%VC 溶液Ⅱ25 μL 并涡旋混匀,添加甲醇500 μL 进行蛋白沉淀,涡旋混合1 min,于6 ℃条件下3000 r·min-1离心10 min,转移上清液150 μL 至装有800 μL 含0.1%1 mol·L-1的乙酸铵水溶液的96 深孔板中,并涡旋混匀,于6 ℃条件下3000 r·min-1离心5 min,进行LC-MS/MS 定量分析。整个制备过程均在冰浴和避光条件下进行。
2.5 标准曲线样本及质控样本的配制和处理
将人空白血浆与20%VC 溶液Ⅰ按照19∶1 的比例进行预处理。将“2.3.2”配制的沙丙蝶呤储备液-Ⅰ用5% VC 溶液稀释,配制成浓度分别为0.02、0.04、0.1、0.2、1、2、4、6 ng·μL-1的沙丙蝶呤系列标准溶液。在预处理过的空白血浆中加入系列标准溶液,配制成浓度分别为1、2、5、10、50、100、200、300 ng·mL-1的标准曲线用标准含药血浆样本。后续处理同“2.4”项。
将“2.3.2”配制的沙丙蝶呤储备液-Ⅱ用5% VC溶液稀释,配制成浓度分别为0.2、4 ng·μL-1的沙丙蝶呤标准溶液。取上述预处理过的空白血浆,配制成浓度分别为1、3、30、150、250 ng·mL-1的质控用标准含药血浆样本,后续处理同“2.4”项。
2.6 临床样本测定
选取10 例健康男性和女性受试者,在健康男性和女性受试者中于空腹情况下评价受试制剂[盐酸沙丙蝶呤片(盐酸沙丙蝶呤,100 mg,口服片剂)]和参比制剂[Kuvan®(盐酸沙丙蝶呤,100 mg,口服片剂)]的吸收速度和程度。受试者将在第一周期中接受受试制剂,然后在至少7 天清洗期后,在第二周期接受参比制剂。在每个周期中,于给药前(-1、-0.5、0 h)和给药后0.5、1、1.5、2、2.5、3、3.5、4、4.5、5、5.5、6、7、8、9、10、12、14、24、36、48 h 共计24 个时间点采集血样,每次取血4 mL,用于血浆中沙丙蝶呤的分析。上述样本处理同“2.4”项。
3 结果
3.1 质谱扫描
在“2.2 质谱条件”下,沙丙蝶呤与沙丙蝶呤-13C15N3主要产生m/z 242.3、m/z 246.1 的[M+H]+峰,选择性地对[M+H]+峰进行产物离子扫描,沙丙蝶呤与沙丙蝶呤-13C15N3的主要离子碎片为m/z 166.1、m/z 170.2。将这些主要碎片离子作为定量分析时检测的产物离子。沙丙蝶呤与沙丙蝶呤-13C15N3的产物离子扫描质谱图见图1。

图1 沙丙蝶呤产物(A)和沙丙蝶呤-13C 15N3 产物(B)离子扫描图
3.2 选择性测试
取6 份不同来源的、经抗氧化剂预处理过的人空白血浆和3 份预处理过的不同来源的空白溶血血浆,除不加内标(改为加同等体积的5%VC 溶液)外,其余按照“2.4”项方法处理。考察空白血浆中内源性物质是否对待测物和内标产生干扰。
沙丙蝶呤和内标沙丙蝶呤-13C15N3的保留时间分别为1.88 min 左右和1.87 min 左右,6 个不同来源的空白血浆中的干扰组分的响应不大于当批次定量下限分析物响应的10.2%,符合小于20.0%的接受标准,内标响应值不大于当批次定量下限内标响应的0.4%,符合小于5.0%的接受标准,表明人血浆中的内源性物质不干扰分析物和内标的定量分析。
3.3 特异性干扰评估
制备定量上限浓度于人血浆空白样本中,除不加内标(改为加同等体积的5%VC 溶液)外,其余按照“2.4”项方法处理,与选择性测试分析批一同制备分析,考察分析物对内标的干扰;另外,在分析批中制备一个空白对照样本(只加内标不加分析物的样本),考察内标对分析物的干扰。
在空白对照样本中,分析物的响应值占当批次定量下限分析物响应值的6.4%,符合小于20.0%的接受标准,表明沙丙蝶呤-13C15N3对沙丙蝶呤样品的干扰可忽略不计;在定量上限浓度样品中,内标的响应值占当批次定量下限内标响应的0.2%,符合小于5.0%的接受标准,表明沙丙蝶呤对内标沙丙蝶呤-13C15N3的干扰可忽略不计。
3.4 标准曲线与定量下限
将“2.5”项下标准曲线用标准含药血浆样本,按照“2.4”项方法处理,以分析物浓度(ng·mL-1)为横坐标X,分析物与内标物峰面积比值为纵坐标Y,用加权(W=1/X2)最小二乘法进行线性回归,标准曲线的回归方程为:Y=aX+b。
沙丙蝶呤的线性范围为1~300 ng·mL-1,典型回归方程为Y=5.72×10-2X+2.57×10-3,r=0.999 9,表明线性关系良好。当沙丙蝶呤定量下限样品浓度为1 ng·mL-1时,其批内与批间平均准确度偏差(RE%)在标示值的±20.0%范围内,变异系数(CV%)在标示值的±20.0%范围内,因此,本分析方法灵敏度达到要求。
3.5 准确度与精密度试验
5 个浓度的质控样本(1、3、30、150、250ng·mL-1),每个浓度分别进行6 重复、6 重复和14 重复,这样连续测定3 个分析批,按照“2.4”项方法处理,使用当批次校准曲线计算各个质控样品的浓度,考察该分析方法的精密度与准确度,结果见表1。表明沙丙蝶呤在各浓度质控样本的批内、批间精密度和准确度符合有关生物样本分析方法的验证要求。
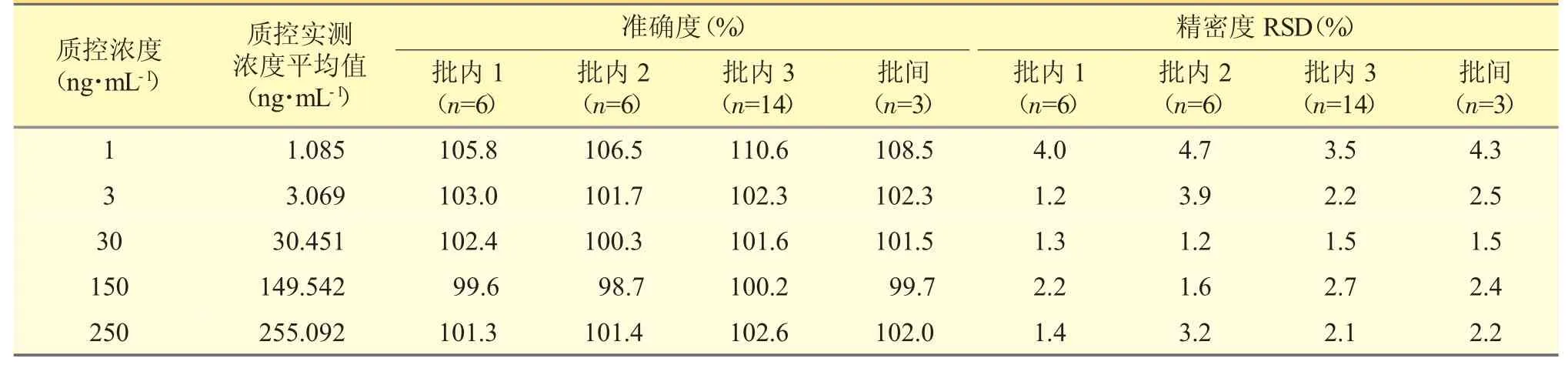
表1 血浆中沙丙蝶呤方法精密度与准确度测定结果
3.6 重新进样试验
考察在“3.5”项中14 重复测定的质控样本,在第一次分析结束后置于自动进样器中91 h(时间的计算:首次进样开始的时间和批次再次进样开始时间的间隔)再次进样,沙丙蝶呤5 个浓度水平的质控样品的准确度平均值在100.2%~104.4%,批内精密度在2.2%~3.9%,结果表明,经处理过的样本在自动进样器内可放置至少91 h,重新进样具有良好的重现性。
3.7 基质效应
考察空白血浆基质效应从低(3 ng·mL-1)、中(150 ng·mL-1)、高(250 ng·mL-1)3 种浓度进行,每个浓度分别用6 个不同来源的空白样品,按样品制备步骤制备后加入含分析物和内标的溶液,配制成相当于低(3 ng·mL-1)、中(150 ng·mL-1)、高(250 ng·mL-1)3 种浓度的样品,即含生物基质的样品。每个样品分析1 次。同时,以纯水为基质,且按样品制备步骤制备后加入含待测物及内标的溶液,配制成相当于低(3 ng·mL-1)、中(150 ng·mL-1)、高(250 ng·mL-1)3 种浓度的样品,即不含生物基质的样品。不含生物基质的样品每个浓度1 个样品,每个样品分析6 次。以此作为对照,考察空白基质效应。
低、中、高3 种浓度的沙丙蝶呤内标归一化基质因子的变异系数(CV%)分别为1.9%、1.3%、1.9%,结果表明,沙丙蝶呤无明显的空白血浆基质效应,不影响待测物的定量分析。
3.8 回收率试验
将“2.3.2”项下方法配制沙丙蝶呤储备液-Ⅰ,用5% VC 溶液稀释,配制成浓度分别为0.06、3、5 ng·μL-1的沙丙蝶呤标准溶液,在100 μL 预处理过的空白血浆中加入5 μL 浓度为0.06、3、5 ng·μL-1沙丙蝶呤标准溶液,分别制备低、中、高3 种浓度的标准含药血浆,每个浓度平行样本3 支,按“2.4”项方法处理,作为回收率试验的提取样本;另外,经预处理过的空白血浆直接按“2.4”项方法处理(不含分析物与内标),经蛋白沉淀步骤后,加入5 μL 浓度为0.06、3、5 ng·μL-1沙丙蝶呤标准溶液和5 μL 的0.2 ng·μL-1内标标准溶液,按配制低、中、高3 种浓度标准含药血浆的量,然后继续制备,每个浓度平行样本3 支,作为回收率试验的未提取样本。提取样本与未提取样本的最后样本组成应相同,于同一分析批中进样分析,以仪器响应值进行评价。沙丙蝶呤的提取回收率公式(同法计算内标):
提取回收率(%)=提取样本的平均响应值(峰面积)/未提取样品的平均响应值(峰面积)×100%
沙丙蝶呤低、中、高3 种浓度的平均提取回收率分别为98.0%、101.2%、98.1%,平均提取回收率为99.1%,回收率的变异系数(CV%)为1.8%;内标的平均提取回收率为92.3%。结果表明,该样品处理方法的回收率较高,不会对样品的分析产生影响。
3.9 稀释测试
用经预处理过的空白基质配制高于高浓度质控样本(250 ng·mL-1)5 倍的样本,将该样本与空白基质以1∶4(高浓度样本∶空白基质)的体积配制,即5 倍稀释,平行操作5 次,将稀释后的样本按“2.4”项方法处理。考察该方法下高浓度样本稀释后是否依然能够准确定量。
高浓度样品稀释5 倍后测定浓度的平均准确度为107.3%,变异系数(CV%)为1.7%,结果表明,对于超过定量上限的样品,进行5 倍稀释后依旧可以准确定量分析,此为待测物浓度超过定量上限样品浓度、但不超过5 倍定量上限浓度的样品进行5倍稀释后的测定提供了依据。
3.10 稳定性试验
考察沙丙蝶呤标准溶液100、6、0.02 ng·μL-1和内标沙丙蝶呤-13C15N3标准溶液50 ng·μL-1、0.2 ng·μL-1,在避光室温放置16 h 和避光放置于-20 ℃冰箱10 d 的稳定性,结果均良好。
用“2.5”项预处理过的空白血浆配制沙丙蝶呤低(3 ng·mL-1)、高(250 ng·mL-1)两种浓度质控血浆样品,分别于避光室温条件以及避光冰浴条件下放置7.5 h、-20 ℃和-80 ℃下反复冻融5 次(解冻条件分为无辅助条件和水浴条件)、-20 ℃下保存7 d 及-80 ℃下保存49 d。按“2.4”项方法处理样本,每一浓度作3 个平行样品,然后进样分析,见表2、表3。用“2.4” 项方法处理后的样本于6 ℃自动进样器存储104 h、在4 ℃冰箱下存储95 h,然后进样,计算浓度,见表4。表明在上述考察条件下,沙丙蝶呤的稳定性良好。

表2 血浆中沙丙蝶呤的短期和长期稳定性(避光,n=3)

表3 血浆中沙丙蝶呤反复冻融5 次的稳定性(避光,n=3)
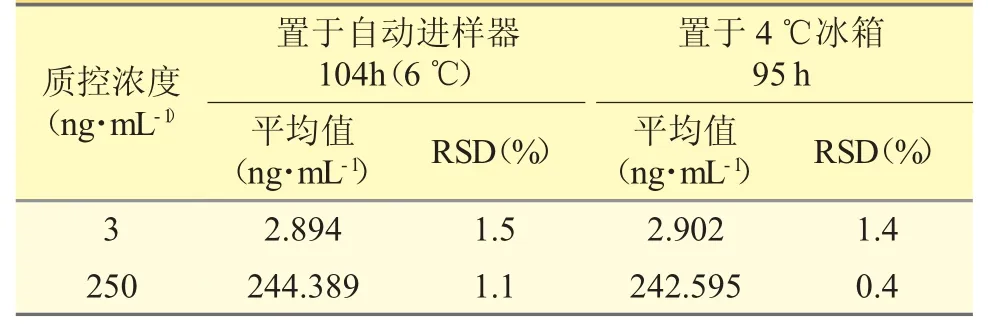
表4 制备后样本稳定性(避光,n=3)
用空白全血,分别配制沙丙蝶呤低(3 ng·mL-1)、高(250 ng·mL-1)两种浓度的全血样品,缓慢混合均匀后立即取出适量,进行低温(4 ℃)离心后,各浓度取3 个血浆样品。同时将剩余全血样品放置于冰浴中,1.5 h 后,将样品再缓慢混合均匀,立即取出适量,进行低温(4 ℃)离心,各浓度取3 个血浆样品。按“2.4”项方法处理后分析,以评价样品采集稳定性。结果表明,1.5 h 平均峰面积比(峰面积比=待测物峰面积/内标峰面积)与0 h 平均峰面积比,差异符合不超过±20.0%的接受标准,表明全血中沙丙蝶呤在冰浴中放置至少1.5 h 的稳定性良好。
3.11 残留测试
残留测试样品为未添加分析物和内标的空白血浆样品,按照“2.4”项方法处理后,续接于校准曲线样品最高浓度后进行分析,考察残留情况,所有分析批均列入考察。结果显示,空白血浆样品在分析物保留时间处的干扰峰的响应值均低于当批次定量下限样本沙丙蝶呤响应值的20.0%,内标保留时间处的干扰峰的响应均低于定量下限样品中内标响应的5.0%,这表明残留可忽略不计,不影响人血浆中沙丙蝶呤浓度的定量分析。
3.12 方法应用
将所建立并已经过验证的方法应用于临床受试者血浆样本的分析。10 例健康受试者口服受试制剂和参比制剂的平均血药浓度-时间曲线图见图2。后续研究将进一步扩大样本量,为沙丙蝶呤的生物等效性研究提供依据。

图2 受试者口服受试制剂和参比制剂的平均血药浓度-时间曲线图
4 讨论
在实验方法建立前,对色谱柱、流动相、内标及离子对的选择等条件进行了摸索,而后选择了最佳的分析方法,并对该方法进行了验证。建立了测定人血浆中沙丙蝶呤浓度的LC-MS/MS 法。
本实验采用QTRAP®6500+型三重四级杆质谱仪,电喷雾电离源(ESI),事实上,沙丙蝶呤在正负离子检测方式下均有响应,但在正离子方式[M+H]+背景下噪音更低,质谱响应信号更强,且干扰少,因此选择正离子检测,沙丙蝶呤在二级扫描中得到主要碎片峰为m/z 166.1。
色谱柱的选择基于实证研究,由于沙丙蝶呤是水溶性物质,经试验,使用色谱柱Kinetex®EVO C18100A˚(4.6 mm×100 mm,5 μm),可以获得较好的峰形,且空白基质背景较低,可以较好地分离分析物和其他干扰杂质。使用该色谱柱平衡时间短,分析时间短,可以满足高通量快速检测的要求,可在最短的运行时间内获得分析物和内标的最佳峰形和质谱响应,具有良好的重现性。选择0.1% 1 mol·L-1乙酸铵的水溶液-乙腈作为流动相,采用等度洗脱的方式,检测物质的峰形良好且能得到最佳的保留时间。另外,由于沙丙蝶呤不稳定,经试验,当柱温高于40 ℃时沙丙蝶呤有明显的氧化反应,因此将柱温设置为30 ℃,保证沙丙蝶呤的稳定性。
选用沙丙蝶呤-13C15N3为内标,其理化性质、质谱响应、色谱行为、提取回收率、基质效应都与沙丙蝶呤相似,且对待测物的测定无干扰,适宜作为本方法的内标。
本方法采用甲醇作为蛋白质沉淀剂,相比于已有文献报道[5-8]方法而言,样本的前处理更简单,有利于大批量制备样本,提高了工作效率。由于沙丙蝶呤的不稳定性会影响到测定的准确性,于是选择在处理过程中分批次加入不同浓度组成的抗氧化剂抗坏血酸(VC),经多次试验,在缺乏抗氧化剂或抗氧化剂浓度较低的情况下均不能准确地测定沙丙蝶呤的浓度,而采用本法测定人血浆中的沙丙蝶呤,其稳定性符合要求。
5 小 结
试验证明,采用本法测定人血浆中沙丙蝶呤的浓度,灵敏度高,专属性强,稳定性好,定量准确,精密度高且易于操作,可实现最低定量下限1 ng·mL-1的测定,分析时间短,分析效率高,可用于大量临床或非临床样本的分析,具有一定的实用性。
