T188K突变遗传型克雅病1例☆
2021-07-12甄超张汉哲张维杰王亚博王新王鸿雁
甄超 张汉哲 张维杰 王亚博王新 王鸿雁
朊蛋白病(prion disease)是一类致死性中枢神经系统退行性疾病,包括克雅病(Creutzfeldt-Jakob disease,CJD)、致死性家族性失眠症(fatal familial insomnia,FFI)、 Gerstmann-Straussler-Scheinker综合征和库鲁病,其中CJD是最常见的一种朊蛋白病。临床上将CJD分为散发型(sporadic CJD,sCJD)、 家族遗传型(familial/genetic CJD,fCJD/gCJD)、医源型及变异型四种。CJD病例罕见,年发病率仅为1/100万~2/100万,确诊的病例中85%~95%均为散发型,遗传型CJD仅占10%~15%。遗传性CJD是由编码朊蛋白的PRNP基因突变所致,目前全球已发现30多种PRNP基因突变可引起遗传型CJD,而T188K位点的突变主要发生于中国汉族人群,且病例数较少。遗传型CJD多表现为快速进展性痴呆、肌阵挛、情绪低落及无动性缄默,伴或不伴有锥体系统/锥体外系统表现,病情进展迅速,多于短期内死亡,但本病早期诊断困难,临床容易误诊,现将我科诊治的1例gCJD报告如下。
1 临床资料
患者,男,63岁,因“睡眠增多伴进行性记忆力减退3个月”于2019年11月10日入院。患者3个月前无明显诱因出现睡眠增多,白天睡眠时间延长,常有困倦感、头部昏沉感,白天可迅速入睡,睡眠中伴喘鸣、肢体肌阵挛,影响日常生活,同时出现记忆力减退,以近记忆力减退为主,伴反应迟钝、言语减少、情绪低落,曾于外院就诊,诊断为“睡眠呼吸暂停综合征”,建议睡眠时佩戴呼吸机辅助呼吸,患者诉佩戴呼吸机过程有呼吸困难、血氧饱和度降低等不耐受情况,未坚持佩戴;患者睡眠增多及记忆力减退情况持续进展,为明确诊断来我院。患者自发病以来,无发热、头晕及意识障碍,无视物障碍、吞咽困难及肢体麻木。饮食欠佳,睡眠增多,大小便正常,体重未见明显变化。既往睡眠良好,睡眠中无打鼾;抑郁病史1年,未规律服用药物治疗;“胆囊切除术”病史。否认重金属、工业毒物及放射性物质接触史,无特殊药物服用史,否认生食牛羊肉及疫区久居史。家族史:父母已故,父亲因“甲状腺癌”去世,母亲因“关节炎并发症”去世,生前均无类似表现。家族中无痴呆病人。患者育有1子,体健。神经系统查体:神清,语利,言语减少,情绪低落,反应迟钝,高级智能减退(简易智力状态检查量表得分MMSE=18分,初中文化),眼球各方向活动自如,无眼震,双侧对光反射灵敏,双侧鼻唇沟对称,伸舌居中,四肢肌力5级,左侧肢体肌张力增高,右侧肌张力正常,双上肢腱反射(+),双下肢腱反射(-),左侧 Babinski's征(+),共济协调,痛温觉对称存在,脑膜刺激征阴性。
辅助检查:血尿常规、血沉、肝肾功能、血脂全套、肿瘤标志物(包括可溶性细胞角蛋白19片段、癌胚抗原、糖类抗原125、总/游离前列腺特异性抗原、糖类抗原242、甲胎蛋白)、甲状腺功能七项(T3、T4、FT3、FT4、TSH、甲状腺过氧化物酶抗体、甲状腺球蛋白抗体)、输血常规(乙肝、丙肝、HIV、梅毒)化验未见明显异常。电解质:Na+128 mmol/L(正常值 137~147 mmol/L),Cl-92 mmol/L(正常值 99~110 mmol/L)。腰椎穿刺术:脑脊液压力90 mmH2O,脑脊液无色透明,白细胞计数为 0,Cl-109 mmol/L(正常值 120~130 mmol/L),脑脊液蛋白 0.427 g/L(正常值 0.2~0.4 g/L),葡萄糖正常,细菌涂片(-),免疫球蛋白正常。血及脑脊液自身免疫性脑炎抗体谱、副肿瘤综合征抗体谱均为阴性。颅脑MRI:尾状核、豆状核均未见异常信号(见图1)。标准多导睡眠监测:重度睡眠呼吸暂停低通气综合征;入睡潜伏期为32.5 min,入睡后觉醒次数为9次,总睡眠时间为287.5 min,睡眠效率为48.4%,整晚睡眠质量较差;睡眠结构紊乱,稳定睡眠占比0.0%,不稳定睡眠占比78.6%,REM期睡眠占比21.4%;夜间中度缺氧,非单纯性鼾症。脑电图:所见α节律差,未见三相波,未见慢波及慢活动,诱发未见明显异常。入院后给予银杏叶、浓氯化钠等治疗。

图1 患者颅脑DWI 尾状核、豆状核均未见异常信号。
出院后患者脑脊液14-3-3蛋白(由中国疾病预防控制中心病毒病预防控制所检测)(-)(图2)。血液PRNP基因(由中国疾病预防控制中心病毒病预防控制所检测):T188K基因突变(图3)。确诊遗传型克雅病。目前患者临床症状进行性加重。

图2 脑脊液中14-3-3蛋白的Western-Blot检测结果。M:标记蛋白;(+):阳性对照。5:此患者蛋白条带。1、2、3、4、6、7、8、9:为同期检测的其他患者蛋白条带。
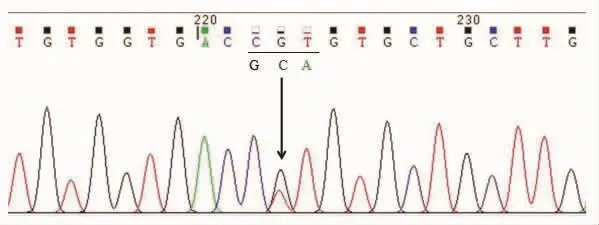
图3 血液PRNP基因Sanger测序(此图展示互补链):PRNP基因563位碱基从胞嘧啶C变成腺嘌呤A(即图中相应互补链从鸟嘌呤G变成胸腺嘧啶T),导致PRP蛋白188位氨基酸由苏氨酸T变为赖氨酸K,即T188K位点杂合突变。
2 讨论
遗传型CJD是由编码朊蛋白的PRNP基因突变所致,PRNP基因位于人类20号染色体上,包含两个外显子,第二个较大的外显子编码253个朊蛋白氨基酸。目前全球已发现30多种PRNP基因突变可引起遗传型CJD,包括点突变或八肽编码区中24 bp的插入突变,相关的点突变包括G114V、V108I、E200K、T183A、T188K 及 I201V 等。而T188K位点的突变主要发生于中国汉族人群,且病例数较少。T188K gCJD是汉族人群中第二常见的遗传性CJD,仅依次FFI;而国外目前仅有 4例病例报告[1-2]。第一例T188K gCJD在2000年由德国学者FINCKH等报告[1]。2009年我国报告了一例T188K gCJD中国患者[3],本病例临床表现与之有诸多相似之处,均以睡眠增多、睡眠结构紊乱起病,常诉头昏、疲劳感,有肌阵挛、言语减少、情绪低落,逐渐出现记忆力减退、痴呆。
T188K gCJD多在50岁以后发病,男性发病率略高于女性,以进展性痴呆、小脑性共济失调、肌阵挛、锥体系统或锥体外系同症状、无动性缄默为主要临床表现,其中2/3患者以进展性痴呆为首发症状,整个临床病程中,93%的患者会发生进展性痴呆,60%患者有肌阵挛,43%患者出现无动性缄默[4]。临床表现与散发性CJD类似,但本病例的突出特征除进展性痴呆外还有睡眠增多及睡眠结构紊乱,恰与FFI的失眠表现相反(其余症状两者多有相似之处),那么睡眠增多同时睡眠结构紊乱是否提示为本病特异性表现,且gCJD与FFI之间是否有重叠交叉,需要更多的病例及检测手段来论证。70%gCJD脑脊液中14-3-3蛋白阳性,阳性率并非100%,有文章指出脑脊液14-3-3蛋白只在疾病初期较短的时间内较易检出[5],因此14-3-3蛋白阴性并不能排除本病。脑MRI表现为尾状核、壳核在DWI呈现对称或不对称性高信号[4]。大部分患者脑电图没有典型的周期性高幅棘-慢综合波(periodicsharp wave complexes,PSWC),且大部分病例无家族史。治疗方面目前尚无特效药物。病情多快速进展,本病预后极差,生存时间明显短于sCJD及FFI,83%患者在症状出现后的5个月内死亡,均位生存时间仅为4个月[4]。
本病例63岁起病,病程快速进展,临床上有睡眠增多、睡眠结构紊乱、快速进展性痴呆、情绪低落、肌阵挛及锥体束征等表现,MRI及脑电图无典型表现,CSF 14-3-3蛋白阴性,基因检测证实PRNP基因T188K突变,明确gCJD诊断。家族中其他成员无痴呆者,父亲因“甲状腺癌”去世,母亲因“关节炎并发症”去世。患者儿子目前无认知功能下降等神经系统表现,因多种原因未行基因检测。本病临床中需要与自身免疫性脑炎、中毒、副肿瘤综合征等疾病相鉴别。
综上所述,gCJD发病率较低,目前临床医生对本病认识尚不充分,极易漏诊误诊。临床工作中遇到50岁以后起病的快速进展性痴呆患者,同时伴有睡眠增多、睡眠结构紊乱、肌阵挛、情绪低落或无动性缄默等表现时应考虑到本病的可能性,即使颅脑MRI、脑电图检查未发现CJD特征性表现,也应进一步送检。
