SACS基因新发突变致遗传痉挛性共济失调1例☆
2021-07-12周燕妮杨文波刘小妮董思其俞海王久存陈向军
周燕妮杨文波 刘小妮 董思其俞海 王久存 陈向军
Charlevoix-Saguenay型常染色体隐性遗传痉挛性共济失调(autosomal recessive spastic ataxia of Charlevoix-Saguenay,ARSACS),是一种遗传性神经退行性疾病。最早由BOUCHARD等[1]于1978年发现于加拿大魁北克省。ARSACS典型的临床表现包括进行性小脑共济失调、周围神经病变伴远端肌肉无力萎缩、下肢痉挛[2]。该病进展缓慢,目前尚无特异性治疗药物,患者多于40岁左右丧失行走能力,50岁左右死亡[3]。2000年,编码 sacsin蛋白的SACS被确定为ARSACS的致病基因[3]。进一步的研究表明,魁北克地区90%以上的患者起病是由SACS基因的6594delT纯合缺失突变引起[4],且患者拥有较为相似的临床表型。后陆续发现SACS新的突变位点,迄今已报告的突变位点有200多种[5-6]。有关ARSACS不同临床表型的报告越来越多,如患者的起病年龄可为婴儿期至40岁以上[5],除了典型症状外,患者还会出现认知损伤、癫痫[7]、骨骼异常和皮肤病变等临床表现[8]。疾病表型异质性较大为临床诊断带来了一定的挑战。本文报告1例由SACS基因复合杂合突变引起的ARSACS,并做文献复习,以增加对该病基因型与临床表型的认识。
1 临床资料
1.1 病史患者,男,34岁,因“双下肢进行性无力伴步态异常11年”于2019年就诊于我院。患者于11年前无明显诱因下开始逐渐出现以近端肌为主的双下肢乏力。症状缓慢进展,于7年前起出现走路拖沓、6年前起自觉下肢僵硬,现尚可独立行走、上下楼梯。病程中伴有双足肌肉萎缩。否认毒物接触、手术外伤、传染病史及冶游史。
生长发育及个人史:足月顺产儿,幼年运动生长发育正常。从小体育成绩尚可。否认父母近亲结婚。父母、妻子、弟弟身体健康。已婚,育有一男一女,健康状况良好。
1.2 神经系统体格检查神志清楚,应答流畅,高级认知功能粗测正常。双眼视力正常,眼球活动自如,未引出眼震。双侧屈髋肌力4级。余下肢肌力正常。双足无畸形,有肌肉萎缩(图1),未见束颤。双上肢肌张力正常,下肢肌张力明显增高。双上肢腱反射未引出,双膝、踝反射(+)。双下肢Babinski’s 征(+)。双侧踝阵挛(+)。颜面、躯干及四肢触觉、针刺疼痛觉对称,四肢振动觉、位置觉、运动觉及皮层觉功能正常。宽基步态,双侧指鼻、跟膝胫试验尚稳准。

图1 小腿肌肉萎缩
1.3 辅助检查血常规、尿常规、粪常规、凝血功能正常。腰穿脑脊液压力不高,常规、生化无殊。
头颅磁共振成像(magnetic resonance imaging,MRI)无明显异常。胸椎MRI示脊髓萎缩伴有中央管扩张(图2)。肌电图示多发性周围神经损害,运动和感觉神经髓鞘及轴索损害。

图2 胸椎MRI提示脊髓萎缩
光相干视网膜断层扫描(optical coherence tomography,OCT)检查示右眼上、颞、下侧视网膜神经纤维层(retinal nerve fibre layer,RNFL)变厚,左眼上、下侧 RNFL 变厚(图3)。

图3 OCT检查结果发现RNFL增厚
1.4 基因检测结果及验证经患者本人及家属知情同意后,抽取患者新鲜外周血进行基因靶向二代测序(杭州迪安医学检验中心)。对遗传性痉挛性截瘫的113个基因和周围神经病的104个基因的外显子及±10 bp内含子区域进行高通量测序,发现患者仅有SARS基因突变并进行Sanger测序法进行验证。结果显示,患者在SARS基因(NCBI reference SACS_ex10 NM_014363.5)上,有两处位点突变:分别为移码突变c.10776delA(p.Lys3592fs)和错义突变c.10796A>C(p.Gln3599Pro)。确认该患者为先证者后,提取其父母新鲜外周血进行家系验证(家系图见图4)。家系验证结果显示,先证者的移码突变来自父亲(p.Lys3592fs),错义突变来自母亲(p.Gln3599Pro)。对其子女的血样也进行了Sanger测序,结果均为c.10776delA(p.Lys3592fs)杂合突变。搜索千人数据库、NCBI数据库(Clinvar、SNP)、ExAC数据库及本地数据库,此两项突变均未见收录及文献报道,两个突变位点在正常东亚人群中的频率也未见报道。错义突变位点用SIFT和Polyphen-2软件对其蛋白保守性进行预测,结果均为有害。根据美国医学遗传学与基因组学学会(American College of medical Genetics and Genomics,ACMG)指南新发突变位点致病性分类标准,患者的移码突变为致病突变(pathogenic),错义突变为疑似致病突变(likely pathogenic),因此认为该患者基因SACS的杂合突变有致病性。

图4 家系图谱
2 讨论
ARSACS是一种遗传性神经退行性疾病,其典型的临床“三联征”表现为小脑性共济失调、痉挛性共济失调步态和周围神经病变[9]。患者头颅MRI在T2和FLAIR上可见小脑萎缩和桥脑线样低信号[10-11]。肌电图检查常有运动、感觉神经传导速度的减慢及复合肌肉动作电位、感觉神经动作电位波幅的降低,提示累及髓鞘和轴索的多发性感觉运动神经病[11-13]。OCT检查常可见患者的RNFL增厚[13-14]。2000年,SACS被确定为ARSACS的致病基因,其位于染色体 13q12.12上,可编码功能未知的 sacsin蛋白[8,14]。sacsin蛋白在神经元中高度表达[8],其功能会随着蛋白结构的变化而改变[15]。进一步研究表明,魁北克地区90%以上患者起病是由SACS基因的6594delT纯合缺失突变引起[4],且患者间拥有较为相似的临床表型,主要表现为典型的“三联征”。由此推测魁北克地区患者间相似的临床表型与其相同的突变位点有关,而非魁北克地区患者在临床表型和突变位点上与魁北克地区患者存在较大的异质性(见表1),提示特定基因突变位点可能与特定表型相关联。
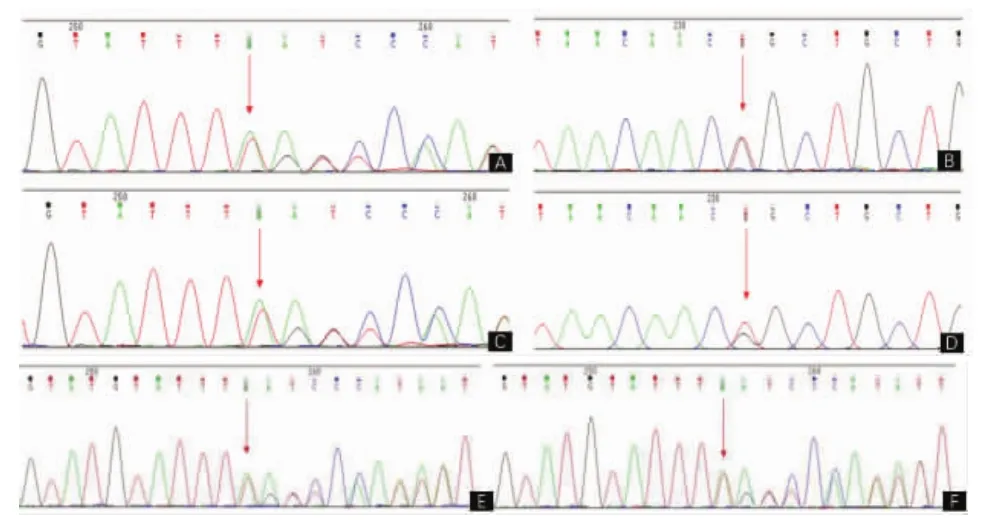
图5 基因测序结果 A.propositus,SACS_ex10 c.10776delA(p.Lys3592fs);B.propositus,SACS_ex10 c.10796A>C(p.Gln3599Pro);C.propositus’s father,SACS_ex10c.10776delA(p.Lys3592fs);D.propositus’s mother,SACS_ex10 c.10796A>C(p.Gln3599Pro);E.propositus’s son,SACS_ex10 c.10776delA(p.Lys3592fs);F.propositus’s daughter,SACS_ex10 c.10776delA(p.Lys3592fs)。
通过高通量测序,我们发现本例患者存在SACS基因复合杂合突变,且两个突变位点均为新发位点。其父母、子女均为杂合突变但无相关临床表现。本例患者SACS基因上的移码突变为致病突变,可能会导致编码蛋白序列提前终止,产生截短蛋白或被降解,对蛋白质的结构和功能可能会产生影响,进而引发疾病;错义突变为疑似致病突变,对对应编码蛋白保守性进行预测也显示其为有害突变。由此推测患者SACS基因上的两个位点突变是引起患者临床表型的主要原因。
由SACS复合杂合突变引起的ARSACS一般于儿童或青少年时期起病[16-17]。而本报告中患者起病较晚,临床表现为双下肢痉挛性截瘫、周围神经损害及躯干共济失调。患者头颅MRI正常,未见小脑萎缩,但其胸部MRI显示下颈部上胸部脊髓变细。肌电图有部分损害,此类变化未有明显外因导致,推测是由SACS基因突变所致。OCT检查显示双眼均有RNFL变厚,这与REZENDE等[6]和LINT等[18]观察到的表型特征一致。
总结既往ARSACS复合杂合突变的病例报告,相关信息汇总于表1。由表可知,15例患者(19例中3例起病年龄未知,1例为本例报告)中14例于儿童期或青少年期发病(14/15)。所有患者均出现了步态变化、共济失调、运动障碍、肢体远端无力,多数患者在报告时已出现小脑、颈髓或胸髓萎缩(18/19)。15例有肌电图检查资料的患者,其检查结果均提示患者存在周围神经病变。多数患者有眼震(16/19)。在进行OCT检查的患者(12例)中7例患者出现了RNFL增厚。19例中11例出现了构音障碍,7例出现高弓足,此外17例中6例出现了智力低下、神经发育迟缓、认知障碍、精神障碍的症状,还有患者出现了癫痫、吞咽困难、指关节弹性过度的症状。其中相同突变者(家系)间的临床症状及检查结果往往极为相似,具体见表1。

表1 ARSACS复合杂合突变患者的基因型及临床表型汇总

续上表
ARSACS患者中,SARS基因突变位点各异,临床表型也存在较大异质性,同一基因上不同突变位点引起的临床表型的各异是否是由不同信号通路或蛋白质功能之间的差异引起,是否影像学或实验室检查特征与特定的临床表现具有相应的关系,仍需进一步的研究。目前中国关于ARSACS的病例报告较少。本例患者的复合突变为新发突变,可为ARSACS中基因与表型关系的研究提供一定线索。
