脊髓小脑性共济失调1型一哈萨克族家系遗传学特征分析☆
2021-06-15马建华雷晶张艳
马建华 雷晶 张艳
脊髓小脑性共济失调1型(spinocerebellar ataxia type 1,SCA1)是一种常染色体显性遗传性神经系统退行性疾病,通常在30~40岁发病。SCA1最主要的病理表现是小脑萎缩,胶质细胞增生和浦肯野细胞严重丢失,伴随着橄榄核、齿状核和脑干第 III、IV、IX、X、XII对脑神经核退行性病变[1]。临床早期表现为进行性小脑性共济失调、构音障碍,吞咽困难,腱反射活跃、眼球震颤、扫视过度[2-3],伴随病程进展出现眼球扫视减慢、上视麻痹、肌张力减低、辨距不良等[4],疾病晚期由于延髓功能障碍导致呼吸衰竭是患者的主要死因[2-3,5]。国内目前报告SCA1家系均为汉族人群。本研究对新疆地区一哈萨克族家系SCA1患者、家系中健康人员和正常对照者进行ATXN1基因CAG三核苷酸重复拷贝数研究,旨在了解哈萨克族SCA1患者及正常人群基因分布特征,为该病的治疗研究提供方向。
1 对象与方法
1.1 研究对象 本研究先证者为 2011年10月就诊于新疆医科大学第一附属医院神经内科临床初步诊断为脊髓小脑性共济失调(SCA)的哈萨克族患者,经家系调查绘制系谱图(见图 1)。该家系均为哈萨克族人,非近亲结婚。SCA患者临床诊断依据 Harding标准[6]。正常对照者为在新疆医科大学第一附属医院神经内科门诊收集的正常哈萨克族健康对照者60名,均无血缘关系且排除神经系统遗传病及家族史。
1.2 临床资料分析 由2位神经内科医师对先证者及部分家庭成员进行详细的病史询问和神经系统专科检查,并详细记录检查结果。
1.3 基因型检测 ①DNA提取:经所有受试者知情同意后,采集该家系临床疑诊 SCAs的42例(9例患者、33例家系“健康”成员)及60名正常对照者的静脉血5 mL于EDTA抗凝管中。按天根基因组DNA提取试剂盒说明书进行操作。紫外分光光度计A260/A280定量,分装后-20℃保存备用。②基因型筛查:参考相关文献进行引物序列[7]的设计,并由由上海生工生物工程股份有限公司合成,包括SCA1-A 5'-AACTGGAAATGTGGACGTA-3'、SCA1-B 5'-CAACATGGGCAGTCTGAG-3',SCA2-A 5'-GCGTGCGAGCCGGTGTAT-3'、SCA2-B 5'-GGACGAGGACGGCGAAGG-3',SCA3-A 5'-CCA GTGACTACTTTGATTCG-3'、SCA3-B 5'-CTTACCT ACATCACTCCCA-3',SCA7-A 5'-TGTTACATTGT AGGAGCGGAA-3'、SCA7-B 5'-CACGACTGTCC CAGCATCACTT-3'和SCA12-A 5'-TGCTGGGAA AGAGTCGTG-3'、SCA12-B 5'-GCCAGCGCACTC ACCCTC-3'共5对引物,行聚合酶链反应、琼脂糖凝胶电泳筛查相应的基因型。③目的基因的扩增和DNA重组:将筛查出的SCA1目的片段切胶回收纯化,将切胶回收DNA产物做为DNA模板,再次行聚合酶链反应及琼脂糖凝胶电泳验证,并与pMD-18T载体连接,转化于大肠杆菌DH5a感受态细胞,对感受态细胞进行培养,取阳性克隆摇菌,抽提质粒,ABI377型全自动 DNA测序仪自动读取数据。本研究中正常对照者及家系中正常者均为纯合子,可将PCR扩增产物直接测序。
2 结果
2.1 SCA1患者临床资料分析 该家系5代共 108人。发病13例,现存患者9例,发病年龄29~56岁,平均 (42.58±8.67)岁,病程7~25年,平均(14.67±5.80)年,家系谱符合常染色体显性遗传模式(图1)。临床主要以行走不稳、构音障碍为突出症状。其他较常见的症状主要包括双眼水平性眼球震颤、吞咽困难、饮水呛咳、下肢痛性痉挛、声音嘶哑和认知功能障碍。

图1 哈萨克族SCA1家系图
2.2 SCA1基因CAG重复序列检测结果 42例家系成员中SCA1基因突变者19例,已发病患者9例,尚未发病但基因检测异常者10例,均是杂合子。5例已发病患者和4例尚未发病但基因检测异常者(见图2),共9例经重组DNA技术成功(见图3)。5例已发病患者ATXN1异常等位基因CAG 重复数目为 41~47 次,平均(44.00±2.23)次,中间无CAT插入;正常等位基因CAG重复数目是 24~27 次,平均(26.20±1.30)次,均有 2 次 CAT插入;4例症状前患者ATXN1异常等位基因CAG重复数目为 40~43 次,平均(41.75±1.26)次,中间无CAT插入;正常等位基因CAG重复数目分别是 26~29 次,平均(27.50±1.73)次,均有 2 次 CAT插入(见表 1)。

表1 哈萨克族SCA1家系患者CAG重复次数与CAT插入次数统计
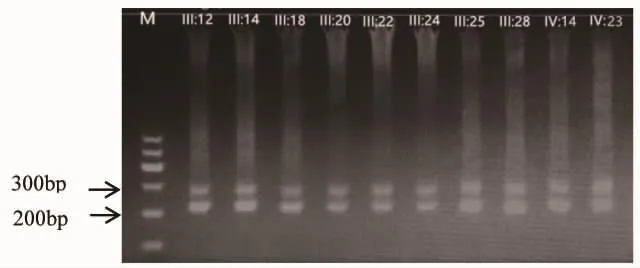
图2 SCA1家系患者PCR扩增后琼脂糖凝胶电泳图 M为marker。
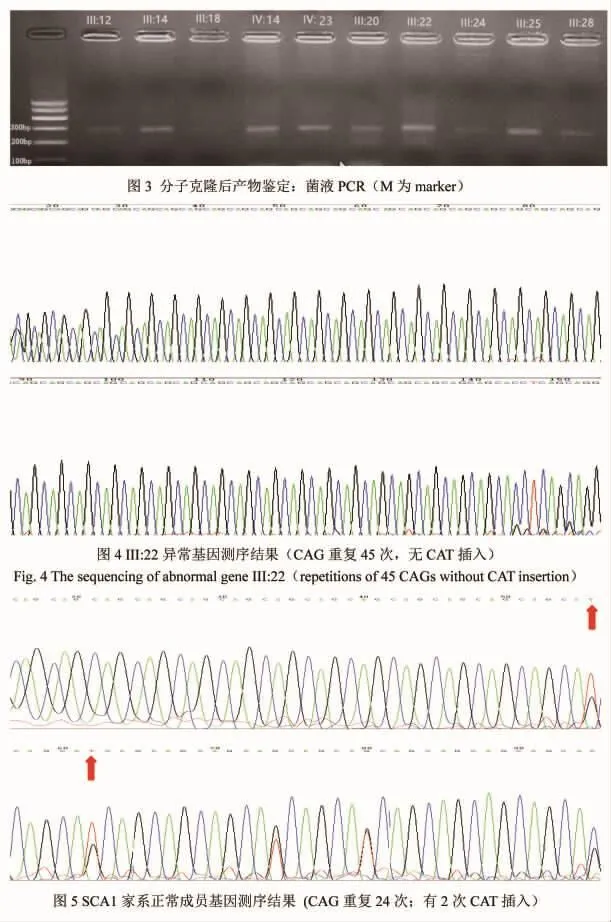
图3 分子克隆后产物鉴定:菌液PCR M为marker。
家系中正常成员共 23名,男10名,女13名,均为纯合子。CAG重复次数范围在17~30次,平均 (25.26±2.90) 次,28次为最常见频率,占21.74%;CAT 插入次数 0~4 次,平均(2.00±1.30)次,2次为最常见频率,占 30.43%(见图 5)。(CAG)nCATCAGCAT(CAG)n 形式出现频率最高,占30.43%。
图4 III:22异常基因测序结果 CAG重复45次,无CAT插入。
图5 SCA1家系正常成员基因测序结果 CAG重复24次;有2次CAT插入。
健康对照者共60名,均为哈萨克族。男25名,女 35名,年龄 14~81岁,平均年龄(46.50±15.31)岁。经检测均为纯合子。CAG重复次数12~30 次,平均(25.87±2.61)次,27 次频率最高占 30%;CAT 插入次数 0~5次,平均(2.10±1.29)次, 2次为最常见频率,占 40.00%(见图 6),以(CAG)nCATCAGCAT(CAG)n形式出现频率最高,占40.00%。
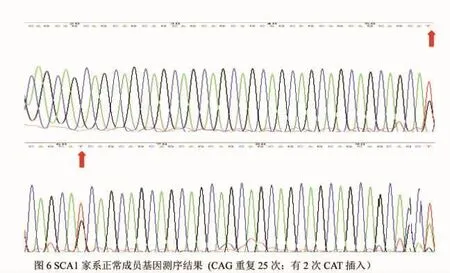
图6 SCA1家系正常成员基因测序结果 CAG重复25次;有2次CAT插入。
3 讨论
SCA1是由位于6p22.3的ATXN1基因第8外显子的CAG三核苷酸序列异常重复扩增,导致其编码的ataxin-1蛋白发生突变。突变的 ataxin-1蛋白主要在Purkinje细胞内形成核内包涵体(INIs),核内包涵体的聚集影响部分细胞内蛋白质的折叠和降解,导致 Purkinje细胞进行性变性[8]。正常等位基因中CAG重复6~35次[9-11],当 CAG连续重复≥39次时,重复次数的大小和发病年龄之间呈负相关。
共济失调是本研究SCA1患者最常见的临床表现,这符合SCA1在宏观上以脊髓-小脑萎缩为主的病理特征。患者均以步态共济失调为首发症状,这与小脑半球病变导致同侧肢体共济失调相一致,而绒球小结节叶和后蚓部的病变通常导致躯干共济失调相一致。
III:12是本研究先证者,40岁发病,主要表现为言语不清,行走不稳等小脑共济失调症状,其异常等位基因CAG连续重复45次。III:9、III:20、III:22分别是先证者大哥和堂哥,其异常等位基因CAG连续重复次数分别为43、44和47次,III:22 CAG异常重复次数虽然大于III:12,但以上三者发病年龄均晚于先证者,鉴于III:12与III:22属旁系而非直系关系,可能与其父系异常等位基因数有关,但其父辈均去世,无法进行验证。III:14是本研究基因重组成功的发病者中唯一一位女性患者,35岁发病,其异常等位基因CAG连续重复41次小于先证者,但其发病年龄却早于先证者,有报告称只有36%~70%的发病年龄差异可以通过CAG重复数目的大小来解释[5],可能存在其他因素影响ATXN1基因产物的表达。本研究中已发病者CAG连续重复次数平均(44.00±2.23)次,与国内韩燕等[12]报告的SCA1家系患者CAG连续重复次数54~68次、唐北沙等[13]报告汉族SCA1先证者及散发者CAG病理重复次数范围在39~60次,以及国外报道的CAG重复次数范围39到>100次[14]相比,哈萨克族人群CAG异常重复次数处于异常值低限。4例症状前患者异常等位基因CAG重复数目为 40~43 次,平均(41.75±1.26)次,因目前尚无临床症状,不能诊断为SCA1,需动态随访观察。
国内姜淼等[15]对东北地区110名汉族正常个体SCA1基因CAG三核苷酸重复序列进行检测,发现等位基因杂合频率84.55%,CAG重复次数在20~39之间,26是最常见重复次数。唐北沙等[16]对300名中国汉族人群SCA内CAG三核苷酸重复序列进行测定,发现等位基因杂合频率为75%,CAG 重复次数在 17~35 次,平均(28.32±2.03)次,28次是最常见重复次数。本研究SCA1正常对照者为哈萨克族人群,均为纯合子,CAG重复次数12~30 次,平均(25.87±2.61)次,27 次频率最高。与前两者比,正常哈萨克族人群CAG重复次数较汉族人群相对偏低,考虑可能与民族差异性存在一定关系。
本研究在进行基因测序时发现正常哈萨克族人群和SCA1患者正常等位基因中存在CAT插入现象,国内对相关报告较少。查阅文献后发现,早在1993年CHUNG等[17]在对126个正常的 SCA1染色体分析中发现98%未扩增的等位基因有一个插入的重复结构CAT,而扩增的30个等位基因中没有该结构的插入。CHUNG等[17]推断SCA1等位基因的扩增可能与CAT插入片段的缺失有关。QUAN等[18]和 FRONTALI等[19]研究发现 CAT在CAG扩增等位基因的插入可以延迟发病年龄,甚至完全消除发病,并由此得出在SCA1等位基因中CAT三核苷酸的插入,抑制了DNA中二级结构滑链结构的形成,防止复制过程中DNA的重复扩增,防止蛋白质错误折叠,提高基因稳定性[20]。这一结论可以解释本研究结果中在正常等位基因中发现CAT结构插入这一现象。欧洲一项关于SCA1的研究也发现ATXN1基因上扩增的和正常的序列均对于预测发病年龄起到很大的作用[21],SCA1患者的发病年龄受ATXN1扩增链和正常链共同影响 ,而且正常链发挥积极的影响 ,这一发现与上述结论不谋而合。所以SCA 1的发病年龄不是由CAG重复的总数决定的,而是由CAG连续重复总数决定[22]。CAG连续重复长度决定了共济失调的严重程度,并且是较好的发病年龄预测因子。
关于SCA异常等位基因在父系和母系传递过程中变化的问题。ORR等[5]和CHUNG等[17]发现63%的父系传递导致CAG重复序列平均扩增+3.3重复,69%的母系传递导致CAG重复序列没有改变或减少;平均而言,母系传递导致重复序列减少-0.4重复。JODICE等[23]发现父系传递导致CAG重复序列增加1.75个,母系的传递导致CAG重复序列减少-0.5个。RANUM等[24]在对SCA1减数分裂不稳定性的研究中得出男性传播占82%,女性占60%。因此CAG不稳定扩增具有性别差异,更容易发生在父系遗传中。这与精子中的SCA1等位基因更不稳定有关,代际间重复扩增可发生于精子细胞前体分裂、精子细胞成熟、减数分裂、减数分裂后成熟和早期胚胎发育中任何一个时期[25]。本研究结果虽然可见代际间存在CAG重复次数波动(III:9、IV:1和V:8是一完整家系,异常CAG重复次数分别为43、43和42次),但没有明显的差异,并且由于本研究未能将患者及症状前患者异常等位基因内CAG重复次数完全检测出来,无法进行父系及母系遗传结果的统计学分析对比,但根据大量研究数据结果看来,仍支持上述观点。
哈萨克族SCA1家系是国内首次报告少数民族家系,与汉族人群相比,其异常等位基因及正常等位基因重复次数均略低,可能与该病存在种族差异性有关。等位基因中CAT结构的插入更有利于基因的稳定性,可能对该病治疗的研究提供帮助。
