系列芳香烃在土壤中洗脱规律的分子动力学模拟
2021-05-26李柏林刘志楠郭书海
李柏林, 刘志楠, 郭书海, 王 丽
(1. 东北石油大学 化学化工学院, 黑龙江 大庆 163318; 2. 中国科学院 沈阳应用生态研究所, 沈阳 110016)
与大气污染物[1]不同, 石油污染土壤中以稠环芳香烃为主要污染物[2], 其中三环和四环芳香烃残留量较多[3], 具有强致癌性、 低挥发性、 低水溶性和低生物降解性, 长期危害土壤生态系统[4]. 以表面活性剂洗脱方法修复芳香烃污染土壤技术是一种经济有效的修复技术[5]. 表面活性剂在油/水界面吸附形成界面膜[6], 可显著改变油/水界面性质[7-8], 该吸附成膜过程存在表面活性剂和油相组分间的相互作用. 土壤中芳香烃结构变化时, 表面活性剂对其进行洗脱的效果有差异, 表明不同苯环数的芳香烃在形成芳香烃/水界面性质不同. Zhou等[9]和Yang等[10]对污染土壤中的菲洗脱, 发现十二烷基苯磺酸钠(SDBS)加强了辛基苯酚乙氧基化物(TX-100)对菲的溶解性; Yang等[11]对土壤中的蒽和芘洗脱, 发现吐温80(Tween80)-皂苷对蒽的洗脱率高于对芘的洗脱率; Yu等[12]对土壤中的菲和芘进行降解, 发现生物表面活性剂浓度增加时, 对菲的降解增强高于对芘的降解增强.
表面活性剂作用于特定的油/水界面产生最大吸附, 油相性质变化导致表面活性剂功效差异化[13]. 文献[14-15]给出了油/水界面性质的分子动力学模拟研究, 表面活性剂分子在油/水界面的计算结果和理论模型高度契合[16]. Lamia等[17]采用透射电子显微镜结合分子动力学模拟的方法, 研究了沥青质为油相时与十二烷基苯磺酸钠间的强静电作用. Ruiz-Morales等[18]采用耗散分子动力学模拟方法, 研究了甲苯为油相时沥青质在油/水界面的取向状态.
芳香烃的毒性随苯环数的增加而增加[19]. 目前的研究中尚未讨论不同环数芳香烃对油/表面活性剂/水界面的性质影响. 本文选用土壤中常见的系列芳香烃为油相, 包括苯、 萘、 菲和芘[20]. 用十六烷基间二甲苯磺酸钠为表面活性剂, 其在土壤中的分解性能力较支链烷基苯磺酸钠高[21]. 与实验研究相比较, 全原子分子动力学方法能在分子水平上提供随着芳香烃环数的增加, 表面活性剂在芳香烃/水界面处的聚集形态及动力学信息. 本文采用全原子分子动力学方法, 从分子水平上研究芳香烃环数变化对芳香烃/水界面性质的影响, 为土壤中芳香烃洗脱规律研究提供理论依据.
1 模型构建与模拟方法
1.1 模型构建
图1为不同苯环数芳香烃相分子(苯、 萘、 菲、 芘)的模型构型, 按苯环数对其简化命名, 记为n-ben(n=1,2,3,4). 计算采用美国Accelrys公司Materials Studio2017软件包中的Focite模块进行分子动力学模拟[22]. 首先用Visualizer模块构建芳香烃相、 水相以及表面活性剂分子模型. 然后用Amorphous Cell工具构建水层、 芳香烃层及单分子层表面活性剂, 其中水层为800个水分子; 单侧芳香烃层为80个芳香烃分子, 芳香烃层共160个芳香烃分子; 表面活性剂层为6个十六烷基间二甲苯磺酸钠分子组成的单分子层, 表面活性剂层共12个表面活性剂分子. 最后用Build Layer工具将各单层组合成芳香烃/表面活性剂/水体系, 所建模型为两侧芳香烃层, 中间水层, 表面活性剂沿垂直界面方向(z轴方向), 亲水基朝向水层, 疏水基朝向芳香烃层. 该模型体系大小为2.9 nm×2.9 nm×17.3 nm.

图1 不同苯环数芳香烃相分子的模型构型
1.2 模拟方法
模拟计算过程均在Forcite模块中完成, 采用COMPASSⅡ力场进行模拟[23], 水分子采用SPC模型. 在建立体系前, 先分别对芳香烃分子、 水分子以及表面活性剂分子采用智能几何优化方法进行30 000步初始化优化, 使分子能量达到最小化, 消除搭建过程中可能导致的分子重叠. 系统搭建完成后, 再对系统进行能量最小化. 优化后的系统先在正则系综(NVT)下进行1 ns的动力学模拟, 然后进行1 ns等温等压(NPT)下的动力学模拟, 使系统达到平衡, 最后进行1 ns的NVT动力学模拟并提取最后500 ps的平衡体系用于芳香烃/水界面分析.
所有模拟过程中的时间步长均为1 fs, 每1 ps记录一次轨迹信息, 初始速率选择随机模式, 温度为318 K, 压力为101 kPa. 用基于原子的方法计算范德华相互作用(vdW)和静电相互作用(electrostatic). 用Andersen法控制温度[24-25], 选择Berendsen法控制压力[26].
2 结果与讨论
2.1 界面性质对增溶的影响
2.1.1 密度分布及界面厚度
由于各系统密度图相似, 因此仅以3-ben系统为例, 绘制芳香烃/表面活性剂/水体系沿z轴方向的密度剖面图如图2所示, 其中(A)为未加入表面活性剂, (B)为已加入表面活性剂. 由图2可见, 平衡后水的密度为(998±25)kg/m3, 其与318 K时纯水的密度990 kg/m3相符, 表明该模拟体系构建正确, 力场参数无误, 可用于芳香烃/水界面性质模拟研究.
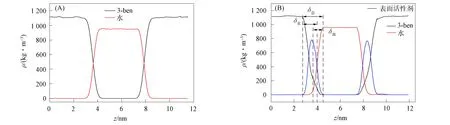
图2 3-ben系统未加表面活性剂(A)和加入表面活性剂(B)沿z轴方向的密度分布
界面厚度指从水相密度的90%到芳香烃相密度的90%或反向间的区域[27-28], 界面厚度包括δ水,δ油及δ表面活性剂三部分, 其关系为
δ表面活性剂=δ总-δ油-δ水,
其中δ水与δ油为10%~90%芳香烃相或水相的厚度,δ总为10%~90%芳香烃相至水相的厚度, 其关系如图2(B)所示. 界面厚度可直观反映界面上表面活性剂吸附的强弱, 系统的界面厚度越大, 表面活性剂的吸附作用越强[29]. 表1为不同芳香烃相分子的界面厚度. 由表1可见, 未加入表面活性剂的芳香烃/水界面厚度较小, 加入表面活性剂后, 芳香烃水界面厚度显著增加, 芳香烃与水界面间形成一定厚度的“过渡层”, 疏水基团和芳香烃分子间的范德华力使其进入芳香烃相, 亲水基团与水分子间的静电作用和氢键作用, 使水分子聚集在其周围. 其中3-ben形成的界面厚度最大且增加幅度也最大, 可见3-ben系统的界面最稳定, 3-ben分子在“过渡层”的界面活性最强.

表1 不同芳香烃相分子的界面厚度(nm)
2.1.2 界面张力
表面活性剂降低芳香烃/水界面张力的能力和芳香烃相性质密切相关. 当界面垂直于z轴时, 界面张力的表达式[30]为

(1)
其中Lzz是沿z轴方向的盒子长度,Pαα(α=x,y,z)是沿α轴方向的压力. 5种芳香烃相的界面张力如图3所示. 苯环数不同的芳香烃相分子和表面活性剂分子间的作用不同, 影响芳香烃/水界面芳香烃分子的构型和表面活性剂的吸附状态. 由图3可见, 3-ben系统的界面张力最低. 3-ben与表面活性剂的作用较强, 表面活性剂的吸附性强, 疏水基最舒展, 在界面处芳香烃分子与疏水基排列紧密, 3-ben在该系统界面处的分子数最多. 2-ben在该系统界面的张力最大, 在该界面处2-ben与表面活性剂接触最疏散, 表面活性剂与3-ben形成的界面最稳定.
2.1.3 界面生成能
界面生成能指系统加入表面活性剂后芳香烃/水界面减少的能量, 该能量与芳香烃分子、 水分子、 表面活性剂分子及分子间存在相互作用关系[31]. 分子模拟过程界面生成能为
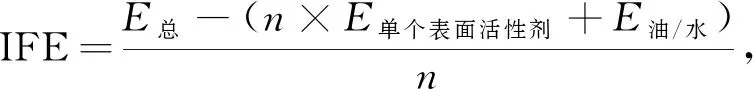
(2)
其中E总为系统的总能量, 当动力学模拟达到平衡时计算得到,E单个表面活性剂为单个表面活性剂分子能量, 在芳香烃/水系统计算时相同条件下得到,E油/水为芳香烃/水界面能量, 在未加入表面活性剂时得到,n=12为表面活性剂分子个数. 界面生成能为负值[32], 其绝对值越大, 芳香烃/水界面越稳定, 界面张力越低. 比较4种不同芳香烃相组成的界面稳定性, 计算各芳香烃相所对应系统的界面生成能(IFE), 计算结果列于表2.

表2 不同芳香烃相组成的界面生成能(kJ/mol)


图3 n-ben(n=0~4)系统的界面张力、 界面厚度(A)和界面生成能(B)
界面生成能增加与界面厚度增加和界面张力降低的变化趋势一致, 模拟计算结果与界面化学理论相符, 3-ben系统界面活性最高、 分子数量最多, 该种表面活性剂对3-ben系统的增溶效果最好.
2.2 芳香烃分子与表面活性剂分子的相互作用
径向分布函数g(r)表示芳香烃相分子在指定半径范围内出现表面活性剂分子烷基链的概率[34], 可反映烷基链的聚集状态与强度. 模型中2,4-二甲基-5-(1′-丁基)-十二烷基苯磺酸钠的丁基和十二烷基分别伸入芳香烃相, 分别对4个模型系统中表面活性剂烷基末端碳原子与芳香烃相分子的径向分布函数进行统计, 计算的径向分布函数结果如图4所示.

图4 十二烷基与芳香烃分子(A)和丁基与芳香烃分子(B)的径向分布函数
由图4可见, 1-ben~4-ben每个系统均出现2个峰, 表明表活性剂分子周围包裹2层芳香烃分子, 超过2个芳香烃分子厚度后, 表面活性剂对芳香烃分子作用消失[35]. 峰间距表示单芳香烃层间距, 强峰表示表面活性剂分子附近芳香烃分子(第一壳层)出现的概率, 弱峰表示表面活性剂分子远端芳香烃分子(第二壳层)出现的概率. 图4(A),(B)中各系统峰间距基本一致, 而峰高不同. 3-ben的强峰最大, 平均值为2.79, 在这4个系统中最大, 因此出现在表面活性剂周围的概率最大, 而且峰间距最短为0.13 nm, 表明3-ben在表面活性剂疏水端形成的芳香烃膜最致密[36].
为进一步研究表面活性剂疏水端附近芳香烃分子的分布情况, 对径向分布函数进行积分, 计算表面活性剂末端2个碳原子与芳香烃分子的配位数[37]:

(3)
其中n为表面活性剂疏水末端周围芳香烃分子的配位数,R为峰的始末,r为芳香烃分子到表面活性剂末端碳原子的距离,ρ为芳香烃分子和末端碳原子的数密度,g(r)为径向分布函数. 计算所得的配位数列于表3.
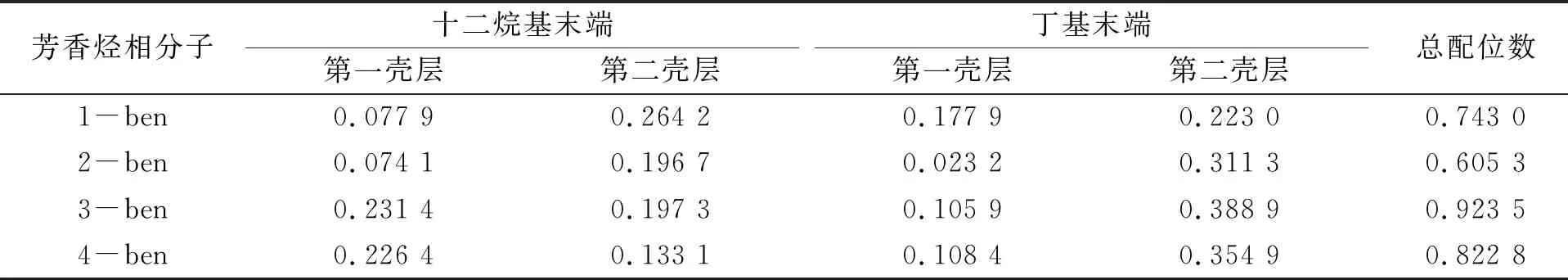
表3 表面活性剂疏水端与芳香烃分子的配位数
总配位数为表面活性剂周围芳香烃分子数, 由表3可见, 1-ben~4-ben配位数大小随芳香烃分子苯环数的变化而变化, 其与界面厚度、 表面张力及界面生成能随芳香烃苯环数变化而改变的趋势一致. 在表面活性剂周围3-ben的总配位数最大, 表面活性剂周围3-ben分子层紧密. 由于3-ben分子之间以及3-ben同表面活性剂之间存在π-π作用和诱导作用较强, 因此使表面活性剂分子疏水端周围容纳更多的3-ben分子, 表现为3-ben系统具有更好的相容性.
2.3 不同芳香烃的洗脱速率
为考察不同芳香烃在表面活性剂作用下的洗脱效率, 对芳香烃相与水相沿z轴方向计算均方位移(MSD)为

(4)
其中〈〉为对系统的所有原子进行平均值计算,r(t)和r(0)分别表示在时间点t和0时的位移. 扩散系数D用均方位移的Einstein方法计算[38]:

(5)
其中N为模拟体系的分子数.
芳香烃相与水相分子的扩散系数列于表4. 由表4可见, 3-ben和水相分子的扩散系数最小, 与1-ben~4-ben在甲醇中扩散系数规律相似[39]. 因为芳香烃/水界面表面活性剂的吸附, 表面活性剂疏水基和芳香烃分子、 亲水基和水分子作用, 3-ben/水界面膜的厚度最大, 界面膜稳定性较强, 所以芳香烃分子向水相迁移、 水分子向芳香烃相迁移能力变弱, 表现为3-ben系统中芳香烃相与水相分子扩散系数均最小, 该系统中3-ben的洗脱速率最慢[40].

表4 芳香烃相与水相分子的扩散系数
3 结 论
本文采用全原子分子动力学方法模拟研究了不同环数芳香烃在以十六烷基间二甲苯磺酸钠为表面活性剂的界面处的分子微观行为, 为洗脱土壤中芳香烃类污染物提供了理论依据, 可得如下结论:
1) 菲/十六烷基间二甲苯磺酸钠/水界面, 具有最大界面厚度、 最大界面生成能及最低界面张力, 增溶效果最好;
2) 菲在十六烷基间二甲苯磺酸钠周围的总配位数最大, 相容性最好;
3) 菲/十六烷基间二甲苯磺酸钠/水系统, 芳香烃相与水相分子的扩散系数均最小, 菲在该系统中的洗脱速率最低.
