团簇Mo2S4的催化活性位点分析
2021-05-09方志刚吕孟娜井润田曾鑫渔
王 倩,方志刚,吕孟娜,井润田,曾鑫渔
(辽宁科技大学 化学工程学院,辽宁 鞍山 114051)
以Pt 为代表的贵金属是性能优良的催化材料,但由于资源短缺、价格昂贵等问题,不能广泛应用于实际生产[1]。过渡金属独特的d 电子结构使其具有独特的物理化学性质。过渡金属原子掺杂非金属原子是有效的催化材料,有望代替Pt 等贵金属催化剂。过渡金属的硫化物[2]、磷化物[3]及碳化物[4]等作为催化材料已经被应用于电池、半导体等领域。非晶态合金具有长程无序短程有序的特点,在催化析氢[5]、磁学性质[6]等方面均有报道。在非晶态Mo-S 体系中,MoS2与Mo6S8都有应用。Cheah 等[7]发现 MoS2具有特殊的二维空间结构,对其光催化性能进行深入探究。Lu 等[8]实验研究发现,使用Mo6S8/C颗粒设计可以转移人工固相电解质界面膜层,提高在碳酸盐电解质中高锂金属的稳定性,使NMC-811 阴极耦合的原型全电池的循环稳定性显著提高。目前,对Mo2S4体系鲜有报道。本文设计团簇Mo2S4初始构型,对其进行量子化学计算,研究团簇Mo2S4的催化活性位点,以期为Mo-S 非晶态合金体系的相关研究提供参考。
1 团簇Mo2S4的计算方法与结构模型
1.1 计算方法
采用与文献[9]相同的量子化学水平B3LYP/Lanl2dz,并根据拓扑学原理[10]设计出 40 种团簇Mo2S4初始构型,在密度泛函理论(Density functional theory,DFT)方法[11]的基础上进行全参数优化计算及虚频验证,排除含虚频的不稳定构型及同重态下相同几何构型中能量较高的构型之后,最终得到单、三重态稳定构型各5种。对Mo、S原子采用含相对论矫正的有效核电势价电子从头计算基组[12],即18-eECP的双ξ基组(3s,3p,3d/2s,2p,2d)优化,并在 S 原子上加极化函数ξS.d=0.55[13]。所有计算均在启天M4390 计算机上利用Gaussian09程序完成[14]。
1.2 团簇Mo2S4的结构模型
团簇Mo2S4优化后的稳定构型如图1所示。右上角标括号内数值表示构型的自旋多重态。构型按几何结构不同可分为:平面六边形(4(1))、五棱锥(4(3),5(3))、四棱双锥(1(1),5(1),1(3),2(3))、带帽三角双锥(2(1),3(3))、三棱柱(3(1))。依据能量参数,设能量最低的构型 1(3)的能量为 0 kJ/mol,计算出其余构型的相对能量,详见表1。
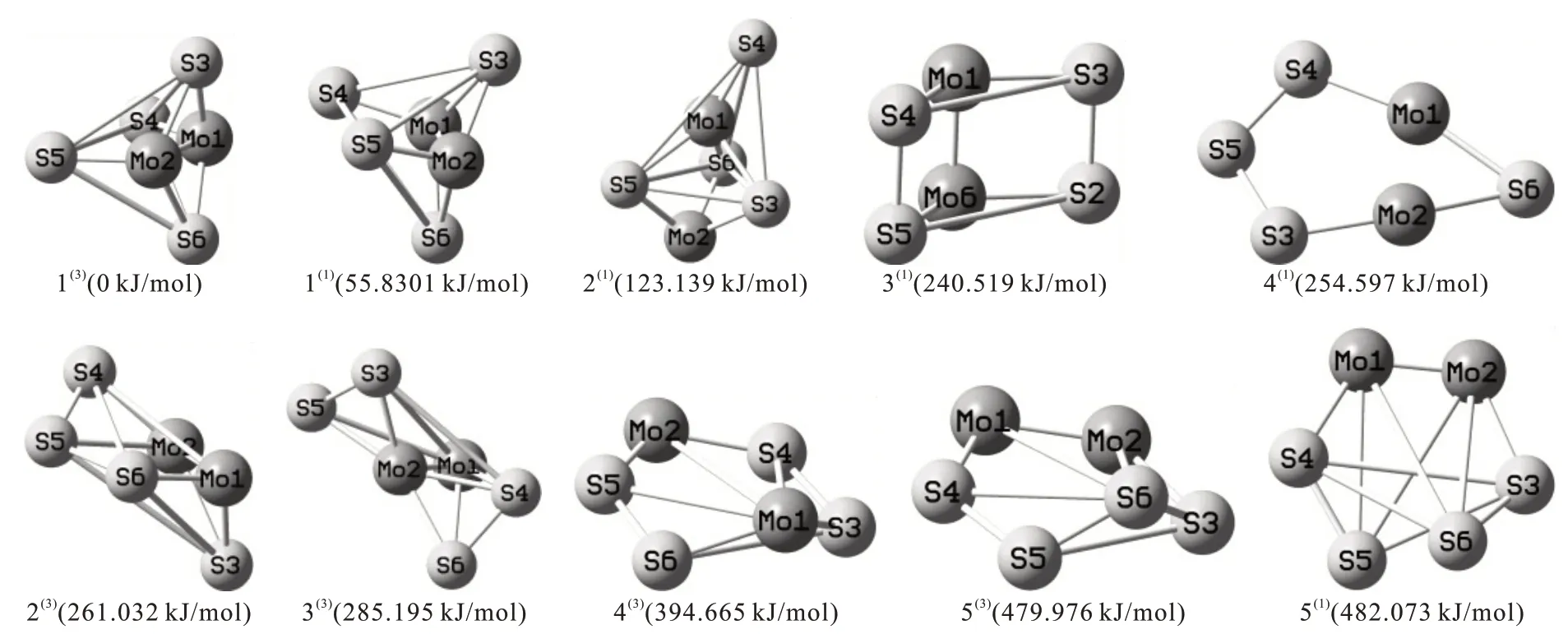
图1 团簇Mo2S4的优化构型图Fig.1 Schematic illustrations of optimized configurations of cluster Mo2S4

表1 团簇Mo2S4的能量参数,kJ/molTab.1 Energy parameters of cluster Mo2S4,kJ/mol
构型能量以及结合能EBE越小,构型越稳定;吉布斯自由能变ΔG越小,反应越容易自发进行。故优化构型的稳定性从高到低排序:1(3)>1(1)>2(1)>3(1)>4(1)>2(3)>3(3)>4(3)>5(3)>5(1)。整体上,单重态的稳定性普遍比三重态的稳定性好。
2 成键分析
原子间键长与键级是判断各原子间成键强度的重要依据。键长越短,原子间重叠程度越大,成键强度越好。键级为负时,不利于两原子间的成键;键级为正时,有利于两原子间的成键。键级值越大,影响成键的能力越大。
表2列出团簇Mo2S4各优化构型中三种化学键平均键长和平均键级。Mo-Mo键的平均键长平均值最小,其成键强度最大,更易成键。Mo-S 键次之,S-S键最弱。Mo-Mo与S-S键级有正有负,Mo-S键级全为正值,说明三种键对构型的作用结果并不单一。除构型2(1)外,其余构型的S-S 键级均为正,表明S-S有成键作用,但S-S键级值较小,对成键的影响较小。Mo-S键级均为正值,有利于成键,对成键的影响强于S-S 键。Mo-Mo 键级有正有负,键级波动范围最大,平均值也最大,对构型的影响最大。
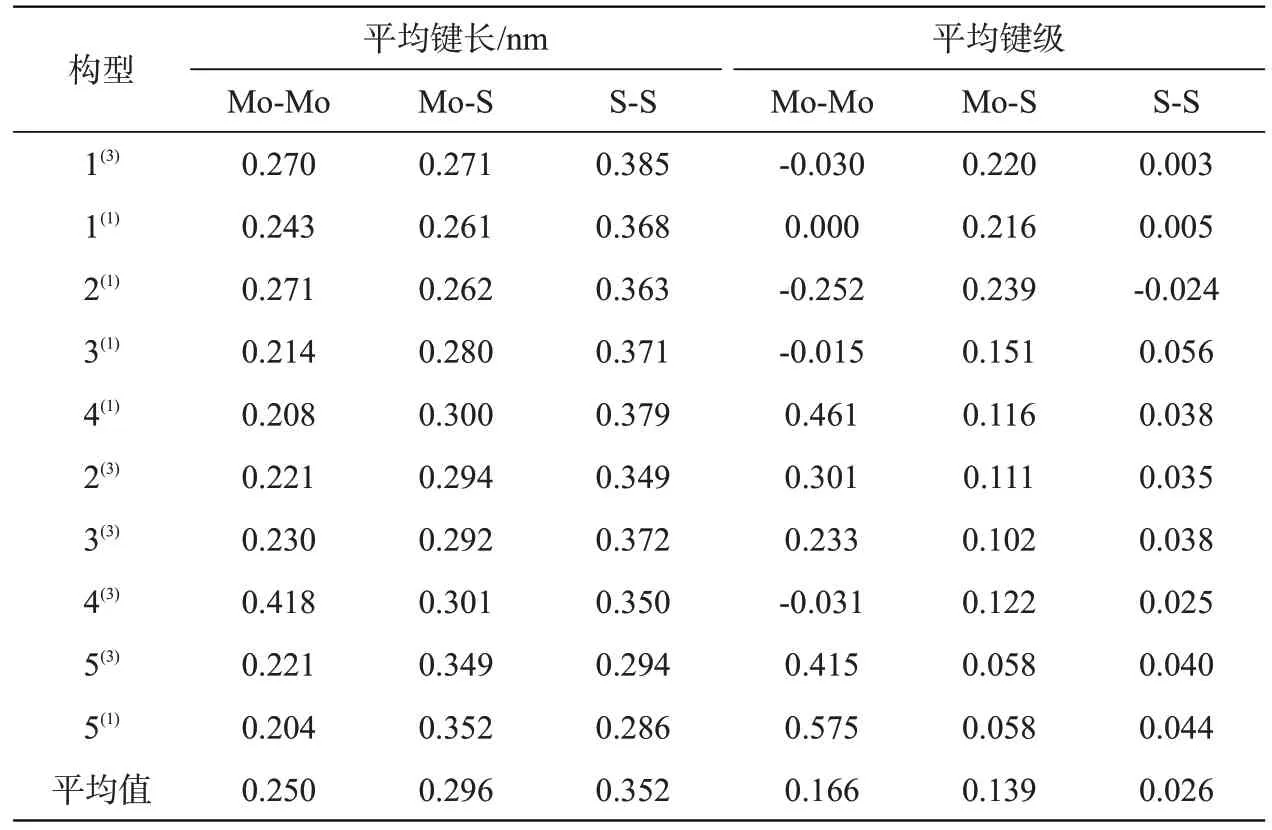
表2 各化学键的平均键长和平均键级Tab.2 Average bond length and average bond order of each chemical bond
综上,三种化学键对构型稳定性的影响排序为:Mo-Mo>Mo-S>S-S。
3 轨道贡献率
最高占据分子轨道(Highest occupied molecular orbital,HOMO)与最低未占据分子轨道(Lowest unoccupied molecular,LUMO)是决定体系中化学反应发生的关键,也是判断团簇分子反应活性的一个重要依据[15]。在理想状态下,假设团簇Mo2S4的形成途径为:2Mo+4S→Mo2S4,Mo 原子和S原子的HOMO轨道中的电子分别流向对方未被电子占据的LUMO 轨道,从而形成化学键,生成Mo2S4。在此过程,Mo 原子和 S 原子在 HOMO、LUMO轨道均有所贡献。
依据轨道贡献率可以确定团簇作为催化剂时所具有的潜在活性位点。将前线轨道能级参数及原子贡献率列于表3 中。其中,EGAP=ELUMOEHOMO,表示各个稳定构型前线轨道的能隙差,能隙差值越小,则在HOMO 与LUMO 之间电子跃迁所消耗的能量越少,团簇化学反应活性越强,说明化学反应速率越快。比较EGAP值,各构型反应活性从高到低排序:5(3)>1(1)>1(3)>2(1)>2(3)>3(3)>5(1)>4(3)>3(1)>4(1)。虽然构型 4(3)和5(3)同为五棱锥几何构型,但因二者原子排布不相同,EGAP值相差较大;而三棱柱构型 3(1)与平面六边形构型 4(1)的EGAP值却相差较小,这说明构型空间结构及原子排布对构型的化学反应活性均有一定程度的影响。此外,除了构型 4(1)、3(3)与 4(3),其余构型中 Mo 原子在 HOMO和LUMO轨道的贡献率均超过S原子,且贡献率均大于50%。这表明Mo原子为团簇Mo2S4前线轨道的主要贡献者,同样也是团簇Mo2S4催化活性中心位点。

表3 团簇Mo2S4的前线轨道能级参数及原子贡献率Tab.3 Frontline orbital energy-level parameters and atomic contribution rates of cluster Mo2S4
为进一步探究催化活性位点的具体位置,将Mo1 和Mo2 原子的贡献率列于表4 中。就平均值而言,Mo2 在单重态和三重态下的HOMO 与LUMO 贡献率均超过Mo1 原子,并且对 HOMO 与LUMO贡献率之和大于50%。这表明在催化反应中,Mo2原子的催化能力强于Mo1。构型3(1)、3(3)、4(1)、4(3)、5(1)和5(3)中,Mo2原子的HOMO 和 LUMO贡献率均超过Mo1 原子,表明Mo2 原子的催化能力强于 Mo1。但在构型 1(1)、1(3)、2(1)和 2(3)中,Mo1原子的HOMO 和LUMO 贡献率之和均超过Mo2,表明Mo1 原子的催化能力强于Mo2。因此,不能忽视Mo1原子的催化能力。

表4 Mo1和Mo2原子贡献率Tab.4 Contribution rates of Mo1 and Mo2 atoms
同为四棱双锥的构型 1(1)和 5(1),虽然几何构型相同,但结合能相差较大,稳定性相差较大。这说明构型的稳定程度、几何结构等因素不仅影响反应发生的难易程度,同样也影响着团簇的催化活性。
4 结 论
基于密度泛函理论,对团簇Mo2S4进行较高量子化学水平的计算,得到10 种稳定构型,其中单、三重态稳定构型各5种。依据团簇Mo2S4的前线轨道能级参数及原子贡献率分析催化活性位点。研究表明,各化学键对构型稳定性的影响排序为:Mo-Mo>Mo-S>S-S;催化反应活性与反应速率排序为:5(3)>1(1)>1(3)>2(1)>2(3)>3(3)>5(1)>4(3)>3(1)>4(1)。在团簇Mo2S4中,Mo 原子为团簇分子的潜在活性位点,对于大部分构型来说,Mo2原子是主要的催化活性位点,但Mo1原子的催化能力也不可忽视;构型的稳定程度、几何结构等因素不仅影响反应发生的难易程度,也同样影响着团簇的催化活性。
