HPLC 法同时测定琥珀酸曲格列汀中9 个杂质的含量
2021-05-08徐佳雯
李 尚,徐佳雯,张 斐,2*
1 徐州医科大学 新药研究与临床药学重点实验室,徐州 221000;2 江苏福锌雨医药科技有限公司,南京 210033
琥珀酸曲格列汀是一种二肽基肽酶Ⅳ(DPP-4)抑制剂[1],通过选择性、持续性抑制DPP-4,进而抑制胰高血糖素样肽-1(GLP-1)和葡萄糖依赖性促胰岛素分泌多肽(GIP)的灭活,提高内源性GLP-1 和GIP 的水平,促进胰岛β 细胞释放胰岛素,同时抑制胰岛α 细胞分泌胰高血糖素,从而提高胰岛素水平,降低血糖[2]。该药是由日本武田公司和Furiex 公司联合研发的全球首个1 周1 次的长效口服降糖药(商品Zafatek)[3],与目前市场上1 日1 次的短效产品相比,有望大幅度改善患者的便利性和依从性,并可潜在提高血糖控制,减少糖尿病并发症。
本研究根据琥珀酸曲格列汀的合成工艺(见图1)、降解途径并结合强制降解试验的结果[4],确定了本品可能存在的杂质有9 个(结构见图2),即杂质Ⅰ~Ⅸ。有关物质是药品质量标准中重要的一项检测指标,通常采用高效液相色谱法(HPLC)进行测定,在各国药典中大多产品质量标准中采用外标法、加校正因子主成分对照法等对杂质进行定量分析,以便确定产品的有关物质变化趋势,判断产品质量。本试验建立了高效液相色谱法同时对琥珀酸曲格列汀及已知的9 个杂质进行分离和含量检测。
1 仪器及药品、试剂
1.1 仪器
Agilent 1260 infinity Ⅱ高效液相色谱仪;Mettler ME55 电子天平(精度0.01 mg)。
1.2 药品与试剂
琥珀酸曲格列汀(含量99.8%,批号20200401、20200402、20200421,江苏福锌雨医药科技有限公司);杂质Ⅰ(纯度97.3%,批号C19005-10-7)、杂质Ⅱ(纯度95.8%,批号C19005-8-3)、杂质Ⅲ(纯度96.8%,批号C19005-13-6)、杂质Ⅳ(纯度97.9%,批号C19005-2-11)、杂质Ⅵ(纯度97.3%,批号190627-2)、杂质Ⅶ(纯度96.1%,批号190705-1),均自制;杂质Ⅴ(纯度96.0%,批号14HW7302-F+1+06940A14)、杂质Ⅷ(纯度95.6%,批号14HW7302-I+3+069481316)、杂质Ⅸ(纯 度97.4%,批 号14HW7302-J+3+069501314)均为南京威诺德医药技术有限公司产品。
磷酸、乙腈为色谱级;盐酸、过氧化氢、氢氧化钠均为分析纯;实验用水为重蒸馏水。
2 方法与结果
2.1 溶液的制备
2.1.1 杂质对照品混合储备液取杂质Ⅰ~Ⅸ对照品适量,加乙腈溶解并稀释制成每毫升中含各杂质100 μg 的溶液。
2.1.2 系统适用性试验溶液取杂质对照品混合储备液和琥珀酸曲格列汀适量,加0.1%磷酸-乙腈(85∶15)溶解并稀释制成每毫升中含各杂质10 μg和琥珀酸曲格列汀500 μg 的溶液。

图1 琥珀酸曲格列汀合成路线图

图2 琥珀酸曲格列汀及其有关物质结构图
2.1.3 供试品溶液称取琥珀酸曲格列汀适量,加0.1%磷酸-乙腈(85∶15)溶解并稀释成每毫升中约含琥珀酸曲格列汀500μg 的溶液,滤过,即得。
2.2 色谱条件
色谱柱:Welch Utimate AQ C18(4.6mm×250mm,5 μm);流动相A:0.1%磷酸溶液,流动相B:乙腈,梯度洗脱:0~3 min、10%B,3~8 min、10%~15%B,8~18 min、15%~25%B,18~20 min、25%B,20~40 min、25%~65%B,40~40.1 min、65%~10%B,40.1~45 min、10%B;柱温:35 ℃;检测波长:230 nm;流速:1.0 mL·min-1;进样量:10 μL。
2.3 方法学验证
2.3.1 系统适用性试验取“2.1.2”项下系统适用性试验溶液,按“2.2”色谱条件进样并记录色谱图,见图3;二极管阵列检测器(DAD)光谱图见图4。在试验结果中,琥珀酸曲格列汀和9 个杂质的分离度均大于1.5,满足分离要求,理论塔板数均大于13000。
2.3.2 破坏性试验精密称定琥珀酸曲格列汀各10 mg(n=5)于20 mL 量瓶内进行强制降解试验:①酸破坏:精密量取0.2 mol·L-1盐酸溶液1 mL,于室温下破坏2 h 后,加入0.1%磷酸-乙腈(85∶15)溶解稀释并定容至刻度,即得;②碱破坏:精密量取0.1 mol·L-1氢氧化钠溶液1 mL,于室温下破坏4 h 后,加入0.1%磷酸-乙腈(85∶15)溶解稀释并定容至刻度,即得;③氧化破坏:精密量取3%过氧化氢溶液1 mL,于室温下避光破坏5 天后,加入0.1%磷酸-乙腈(85∶15)溶解稀释并定容至刻度,即得;④高温破坏:取样品适量,于60 ℃条件下破坏5 天后,加入0.1%磷酸-乙腈(85∶15)溶解稀释并定容至刻度,即得;⑤光照破坏:取样品适量,于(4500±500)Lx 条件下破坏5 天后,加入0.1%磷酸-乙腈(85∶15)溶解稀释并定容至刻度,即得。分别取上述破坏溶液,按“2.2”项下色谱条件进样测定并记录色谱图,见图5。
结果表明,各降解产物峰与主成分峰均分离良好,分离度均大于1.5,且空白溶剂对杂质检出没有干扰,表明该法专属性良好,同时对样品进行物料平衡计算,各条件下破坏样品的总峰面积均在95.0%~105.0%,物料基本守恒,试验数据见表1。
2.3.3 线性关系考察及校正因子测定称取杂质Ⅰ~Ⅸ和琥珀酸曲格列汀适量,加0.1%磷酸-乙腈(85∶15)稀释成浓度分别为0.05、0.1、0.2、0.5、1.0、5.0、8.0、10.0 μg·mL-1的单一成分溶液,取上述溶液,按“2.2”项下色谱条件进样测定,记录峰面积,以质量浓度X(μg·mL-1)为横坐标,峰面积Y 为纵坐标,绘制标准曲线,得回归方程,用琥珀酸曲格列汀线性回归方程的斜率与各杂质线性回归方程斜率比值计算各杂质的校正因子,见表2。
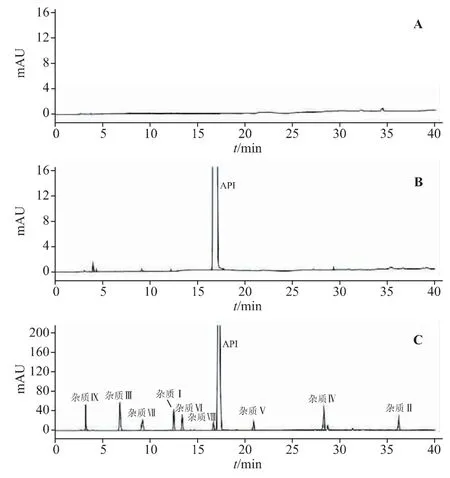
图3 系统适用性试验色谱图
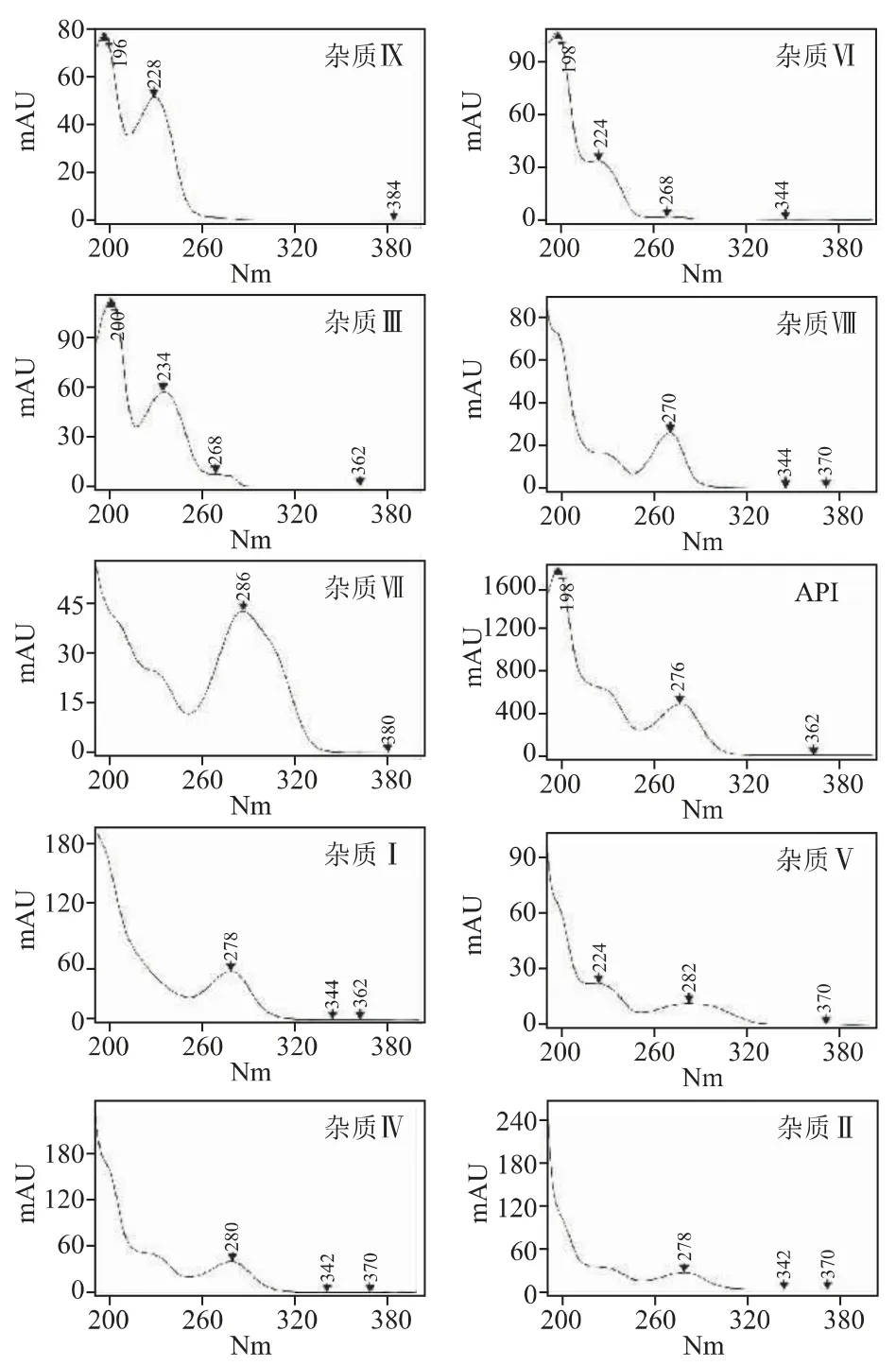
图4 二极管阵列检测器光谱图

图5 专属性试验HPLC 色谱图
结果表明,杂质Ⅰ~Ⅸ和琥珀酸曲格列汀线性关系均良好,r2≥0.9999;杂质Ⅱ、Ⅵ、Ⅸ的校正因子在0.9~1.1,测定杂质含量时可采用不加校正因子的主成分对照法;而杂质Ⅰ、Ⅲ、Ⅳ、Ⅴ、Ⅶ、Ⅷ在测定含量时则需要使用校正因子进行校正。
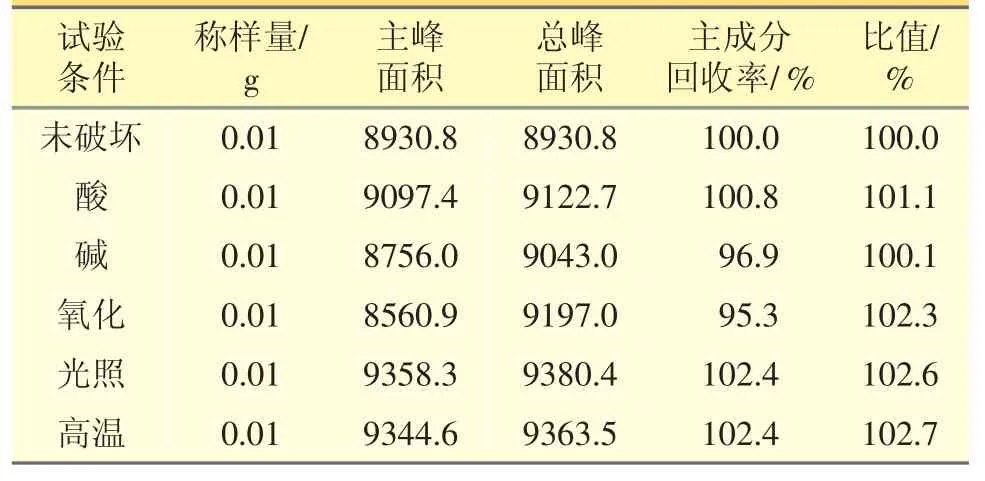
表1 物料平衡数据
2.3.4 检测限及定量限测定取“1.2”项下系统适用性试验溶液,加0.1%磷酸-乙腈(85∶15)逐级稀释后,按“2.2”项下色谱条件进样测定,按信噪比S/N=3 定为检测限,S/N=10 定为定量限。见表2。

表2 琥珀酸曲格列汀与杂质Ⅰ~Ⅸ的线性关系、校正因子、检测限及定量限
2.3.5 精密度试验
进样精密度:按照“2.1.2”项下方法配制系统适用性试验溶液,按“2.2”项下色谱条件进样测定,连续进样6 次,记录峰面积。试验表明,琥珀酸曲格列汀和杂质Ⅰ~Ⅸ峰面积的RSD 值分别为0.1%、0.1%、0.1%、0.2%、0.2%、0.4%、0.1%、0.3%、0.2%、0.9%,结果均<1.0%,表明仪器精密度良好。
重复性试验:取本品,按照“2.1.2”项下方法,平行配制6 份系统适用性试验溶液,照“2.2”项下色谱条件进样测定,利用加校正因子的主成分对照法[5]分别计算各杂质的含量,结果表明,6 份样品的杂质含量基本一致,各杂质和总杂RSD 值均在1.0%以内,表明本方法重复性良好。
2.3.6 回收率试验按照“2.1.3”项下方法配制9份供试品溶液,精密量取“2.1.1”项下杂质对照品混合储备液1、2、3 mL 各3 份,分别加入到上述9 份供试品溶液中,加适量0.1%磷酸-乙腈(85∶15)溶解并稀释,配制成约为已知杂质限度的50%、100%、150%的供试溶液。按“2.2”项下色谱条件进样测定,记录峰面积,通过外标法计算出每份样品中各杂质的测得量,根据测得量与加入量的比值计算每个杂质的回收率,并计算出每个杂质的RSD 值。试验结果表明,杂质Ⅰ~Ⅸ的平均回收率和RSD 分别为Ⅰ:99.6%、0.8%;Ⅱ:99.8%、1.9%;Ⅲ:99.9%、0.3%;Ⅳ:99.6%、0.7%;Ⅴ:100.2%、0.9%;Ⅵ:100.5%、0.9%;Ⅶ:99.4%、1.0%;Ⅷ:100.5%、0.4%;Ⅸ:99.2%、1.4%。均在99.0%~101.0%、均<2.0%,表明准确度较高。
2.3.7 稳定性试验称取琥珀酸曲格列汀25 mg,置50 mL 量瓶中,再向其中精密量取“1.2.1”项下杂质对照品混合储备液5 mL,加入适量0.1%磷酸-乙腈(85∶15)溶解并稀释,配制成溶液,室温放置,分别于0、2、4、8、12 h 后,按“2.2”色谱条件进样测定,记录原料药与各杂质的峰面积。试验表明,琥珀酸曲格列汀和杂质Ⅰ~Ⅸ的待测溶液在室温下12 h 内RSD均<2.0%,表明溶液稳定性良好。
2.3.8 耐用性试验取本品系统适用性试验溶液,分别考察在不同流速(1.0 mL·min-1±0.1 mL·min-1)和不同柱温(35 ℃±5 ℃)的条件下,主峰和相邻杂质的分离度及杂质含量的变化情况。由耐用性试验看出,在本方法流速和柱温发生微小变动的情况下,除保留时间略有变化外,对本品有关物质的检测基本无影响,系统适用性试验溶液各相邻峰之间最小分离度均>1.5,杂质Ⅰ、Ⅱ、Ⅳ、Ⅴ、Ⅵ、Ⅶ、Ⅷ、Ⅸ的RSD 值均<5.0%(含量在0.5%~2.0%),杂质ⅢRSD值<2.0%(含量>2.0%)。结果表明,本法测定琥珀酸曲格列汀有关物质的耐用性良好。
2.4 有关物质检测
取3 批琥珀酸曲格列汀,按照“2.1.3”项下方法配制供试品溶液,按照“2.2”项下色谱条件进样测定,以加校正因子的主成分对照法[5]计算各杂质含量,见表3。
3 讨论
色谱柱的选择:琥珀酸曲格列汀化学结构中含有胺基、内酰胺、氰基等多种含氮基团,是一种碱性化合物;另外,作为一种盐类物质,它具有较强的水溶性,使用常规反相色谱柱分离效果不佳,因此应选择适合能分离碱性化合物并且耐受高比例水相的色谱柱。Welch Utimate AQ C18(4.6 mm×250 mm,5 μm),是一款耐100%水相的色谱柱,对于高含水量流动相具有很好的兼容性,同时该色谱柱采用双封端技术,对于比较难分离的碱性化合物也可获得良好的峰型,因此,最后确定选择该色谱柱。

表3 有关物质含量测定结果(%)
流动相的选择:该药pKa=8.6,在pH>pKa+2 和pH<pKa-2 的条件下,可以保证有良好的峰型,根据色谱柱的适用pH 范围,选择使用酸性体系。由于该药在乙腈中的溶解度较高,故选择其为有机相;在选择水相的过程中,使用10 mmol·L-1磷酸盐(磷酸调pH=3.0)-乙腈和10 mmol·L-1醋酸盐(醋酸调pH=3.0)-乙腈体系时,前者会导致杂质Ⅸ的峰型不佳,后者基线不稳,影响检测,而0.1%磷酸水溶液则可以确保杂质有良好的峰型且基线平稳。综合考虑,最后选择使用0.1%磷酸水溶液-乙腈体系。
检测波长的选择:通过二极管阵列检测器全波长扫描结果(图4)可知,琥珀酸曲格列汀原料药和杂质Ⅰ~Ⅸ在230 nm 波长附近均有较强的紫外吸收,故选择230 nm 作为本品有关物质的检测波长。
强制降解实验:本品在光照、酸性、高温条件下相对来说较稳定,在碱性及氧化条件下结构均不稳定,易形成开环物,且随反应条件加剧,杂质增长明显。本试验使用系统适用性溶液对杂质进行定位,可以初步确定本品在碱性条件下可产生杂质Ⅰ、Ⅲ、Ⅶ、Ⅸ;在氧化条件下产生杂质Ⅰ、Ⅲ、Ⅵ。为了能够更好地控制琥珀酸曲格列汀的质量,可以对降解实验产生的主要杂质进行结构确证。
综上所述,本试验建立了一种高效、专属性强的琥珀酸曲格列汀有关物质HPLC 法。该法可以有效分离主成分和已知杂质,可用于同时测定该原料药中9 个杂质的含量。
