连续微反应加氢技术在脱保护反应中的应用
2021-03-06娄锋炎尹佳滨段笑南王祁宁艾宁张吉松
娄锋炎,尹佳滨,段笑南,王祁宁,艾宁,张吉松
(1 浙江省生物燃料利用技术研究重点实验室,浙江杭州310014; 2 化学工程联合国家重点实验室,清华大学化学工程系,北京100084; 3 浙江工业大学化学工程学院,浙江杭州310014; 4 嘉兴学院生物与化学工程学院,浙江嘉兴314001)
引 言
在医药与精细化工领域中,保护与脱保护是一种较为常见的有机合成策略。多官能团底物进行多步有机合成时,通常需在反应活性位引入相应保护基以避免副产物的生成。常见的保护基主要有苄基和苄氧羰基等。其中N−和O−苄基是有机合成中最常用的保护基[1],一般可通过苄基卤取代或苯甲醛缩合反应将其引入底物分子中[2],用于保护醇、酚、羧酸和酰胺等物质[3],使氨基、羟基等敏感基团在多步合成过程中保持稳定,而后根据产品要求进行苄基脱保护。苄氧羰基是一种常见的胺保护基,又称Cbz[4],主要用于合成多肽。其与苄基保护基均可通过催化加氢、催化氢转移氢解[5−8]、均相还原反应[9]或酶促法[10]等方式实现快速而高选择性地脱保护。当底物分子无酸敏感基团时,还可通过对甲苯磺酸[11]、HBr/HOAc、液态HBr、液态HF[12]和三氟乙酸/茴香硫醚[13]等均相酸实现苄基或苄氧羰基的脱保护。其中,催化氢转移氢解和均相还原法具有反应条件温和、选择性高和操作简单等优势,但存在成本较高、废液产生较多等问题;酶促法面临的菌种筛选和培育问题制约其发展;而催化氢解凭借其原子经济性与高效绿色化等优点在医药中间体等有机合成中得到广泛应用。莫西沙星[14−15]、美罗培南[16]和多利培南[17]等抗菌药的合成过程中均有涉及催化加氢脱保护。
传统的催化加氢脱保护反应[16,18−21]一般选择配有搅拌桨的高压间歇加氢釜为反应器,氢气以气泡形式引入反应体系。搅拌可增强气液两相的湍动程度,增大催化剂表面物质的更新速率;同时破碎气泡使气液两相界面积增大,并延长氢气在液相中的停留时间以增加氢气的溶解量,强化传质和传热效率,但依然存在气液传质效率低、反应时间长等问题,有些较难脱除的苄基甚至需要72 h 才能完成[22]。连续微反应加氢技术是连续流动化学与微反应器技术的结合体,它的出现为实现高效、绿色且可持续的有机化学合成提供了可能[23]。该技术利用微通道的优势增加气液固三相界面接触面积,极大强化了多相传质和传热,并显著缩短反应时间到分钟级甚至秒级。目前连续流加氢反应器主要有壁载式、填充式和浆料式三种路线[24]。文献中报道的连续流脱保护反应基本选择填充式连续流微反应器(图1),其轴向返混通常较小,使气液两相的停留时间分布较窄,可减少连串副反应[25]。其次,由于微反应器体积较小,其氢气滞留量相对较少[26],且催化剂因固载化而无须分离,操作安全性较高。近年来,连续流非均相加氢技术因其安全、高效等特点已得到越来越多的关注。不少国内外学者参与该技术的研究与讨论,并在有关综述[24,26−27]中介绍连续微反应加氢技术在N−、O−苄基和N−Cbz 脱保护方面的应用及其显著优势,包括产物的高选择性和较短的反应时间。
本文首先简要介绍了加氢脱保护中催化剂与溶剂的选择,而后介绍了连续流非均相加氢技术在脱保护领域中的应用,尤其是在药物中间体的合成领域,最后对该技术在脱保护中的应用进行了展望。

图1 用于加氢脱保护的连续微填充床反应器示意图Fig.1 Schematic diagram of continuous micro packed bed reactor for hydrodeprotection
1 催化剂与溶剂的选择
1.1 催化剂对加氢脱保护反应的影响
催化剂对非均相催化加氢脱保护工艺至关重要,尤其是在连续化生产过程中,固体催化剂的稳定性对整个工艺过程有很大影响[28]。其中较为常见的加氢脱保护催化剂如表1所示。
相比于金属镍、贵金属铂、铑和钌,金属钯因吸氢性能好[42]、催化活性高、产物选择性好等优势已成为主流的脱苄基催化剂,也是目前苄氧羰基脱保护的首选催化剂。考虑其成本较高,通常选择比表面积较大的多孔活性炭或氧化铝为催化剂载体,使其催化活性位点得到充分利用。其中活性炭对金属钯的作用力较弱,可减小其对催化剂活性的影响,使Pd/C催化剂具有较好的催化活性[43]。

表1 常见的加氢脱保护催化剂、溶剂和添加剂Table 1 Common hydrogenation deprotection catalysts,solvents and additives
催化剂的加氢活性还与载体孔径分布有关。当底物分子较大时,其无法扩散至载体的微孔和部分中孔[32],且可能堵塞载体孔道,减少催化活性中心的有效利用率。此时,要求载体具有较大的孔径与孔容,使大分子底物能够更好地接触催化活性位。陈莉[16]采用介孔活性炭和普通活性炭负载的钯催化剂进行保护美罗培南的脱苄基/苄氧羰基,研究结果表明前者的加氢活性与稳定性皆明显优于后者,这主要是由于普通活性炭载体上的部分微孔或中孔被大分子副产物阻塞。李岳锋等[44]采用Na2CO3调节催化剂前体H2PdCl4水溶液的pH,使之形成体积较大的钯螯合离子,并借助位阻效应和活性炭表面的丰富基团,使钯螯合离子在浸渍的过程中均匀吸附于活性炭外表面,形成“蛋壳形”Pd/C 催化剂以氢解保护美罗培南。该制备方法既可满足反应物与催化剂活性位的良好吸附,又可消除内部传质阻力的影响。
氢氧化钯含量为20%的Pearlman 催化剂是一种常用于脱苄基的活泼催化剂[1],当Pd/C 催化剂对脱N−苄基失效时,该催化剂仍可显现优越的性能[45]。Bernotas 等[46]还发现该催化剂可选择性使胺加氢脱N−苄基而使苄基醚保持稳定。其认为含胺的底物可有效除去促进苄基醚氢解的微量酸,从而阻碍或抑制苄基醚的脱保护。但由于Pearlman 催化剂的活性组分负载量较高,导致其生产成本较高,一定程度抑制了其应用。Li 等[47]利用1∶1 的Pd/C 和Pd(OH)2/C 混合催化剂对多种脂肪醚/胺与芳香醚/胺进行加氢脱保护,发现等质量的混合催化剂的催化效率较单一的Pd/C 或Pd(OH)2/C 要高,反应时间也可缩减一半左右,并能有效解决Pd(OH)2/C催化剂的成本问题。除Pd/C、Pd(OH)2/C等高活性氢解催化剂外,贵金属/树脂催化剂的研究也得到广泛关注。Dong 等[36]在室温、常压条件下,将10% Pd/C 替换为Pd/Dowex,并利用钯与酸性树脂的协同作用对四乙酰基二苄基六氮杂异伍兹烷进行N−苄基脱保护,产物的收率由92%提升至98%,并减少了贵金属钯的负载量。Koskin 等[48]推测HBIW 氢解期间的催化剂失活由低聚产物堵塞活性炭孔道和金属颗粒团聚引起。Fotouhi−Far 等[49]进一步研究发现催化剂活性位周围的pH 下降会导致部分金属钯晶粒浸入液相,导致催化剂负载量减少,且钯晶粒的团聚将显著减小催化剂比表面积,削弱催化剂对H2的化学吸附作用,从而影响其加氢活性。Maksimowski等[50]为解决HBIW 脱苄基时的Pd/C 失活问题,开发了一种催化剂再生工艺,即在5 L/h 的氮气/水蒸气流中,将失活的催化剂加热至350℃,并持续2 h。使用再生的催化剂进行HBIW 加氢脱苄基,产物收率约为42%。
1.2 溶剂对加氢脱保护反应的影响
溶剂对非均相催化加氢体系的影响也较大,通常涉及氢气的溶解度,溶剂分子在催化剂活性位上的竞争性吸附,催化剂颗粒的团聚以及反应物或产物分子与溶剂之间的相互作用[51]。不同催化剂−溶剂组合的加氢活性略有差异,其中较为常见的加氢脱保护溶剂如表1所示。
连续流催化加氢脱保护通常选择低级醇、四氢呋喃、乙酸乙酯、乙酸和二甲基甲酰胺等极性溶剂为反应溶剂,其中二甲基酰胺在含OH−的水溶液中会逐渐分解为甲酸根与二甲胺[52],而游离胺的存在会影响催化剂的部分活性。鉴于甲醇和乙醇的低毒性、低成本和高溶解性,其通常被视为最佳溶剂。Pandarus 等[40]选取SiliaCat Pd(0)为催化剂,并以甲醇、乙醇或四氢呋喃为反应溶剂分别进行1−(苄氧基)−4−甲氧基苯的加氢脱保护。研究结果表明,底物在甲醇中的氢解速率最快,且底物浓度过高会减缓甚至抑制氢解反应。
脱保护溶剂还可分为单相和双相溶剂,后者有助于实现氢解产物与保护基的相分离,可有效解决目标产物的分离问题[17],并避免因有机组分快速固化而造成管线堵塞[29]。Perosa 等[34]分别利用纯乙醇和KOH−异辛烷−Aliquat®336 混合溶剂进行苄甲基醚的氢解,发现Pd/C 催化剂在乙醇中的氢解效率明显较高,而Raney−Ni 恰恰相反。Perosa 等推测催化剂表面涂覆的Aliquat®336 可促进Raney−Ni 向反应的有机相转移,并为底物提供具有特定官能团催化位点的反应微环境。
David 等[19]为研究体系pH 与脱苄基和脱氯的选择性关系,以4−氯−N,N−二苄基苯胺为底物,并采用三乙胺/乙酸缓冲液调节反应体系的pH。研究结果表明,当体系的pH 高于被保护胺的pKa时,可以实现高选择性脱氯;反之,有机胺将以质子化形式存在,增强N−Bn 键(Bn 代表苄基)的亲电性,既可抑制脱氯的部分活性,又能防止催化剂失活。
本课题组[33]在连续氢解苄基类保护基时发现四氢呋喃和2−甲基四氢呋喃对Pd(OH)2/Al2O3催化剂具有一定的毒害作用,可使氢解转化率缓慢下降,但具体的催化剂失活机理有待进一步研究。
此外,氢气在传统有机溶剂中的溶解度一般较低,但与超临界流体(如超临界二氧化碳)完全混溶。超临界流体凭借其低黏性、良好的传质和传热性能以及易与产物和溶剂相分离,将在连续催化加氢领域占据重要地位[53]。
2 连续微反应加氢脱保护
2.1 脱苄基反应
2.1.1 O−苄基脱保护 O−苄基是药物合成过程中的常用保护基,可用于保护酚、醇和羧酸类有机化合物。在有关药物化学的文献资料中,O−苄基的脱除占所有脱保护反应的1.4%[54]。多羟基化合物的合成过程中,各保护基的氢解顺序会严重影响加氢产物的选择性。因此,需要不同反应活性的苄基保护基实现正交保护基策略[55]。Gaunt 等[55]为此展开相关研究,发现不同苄基醚的氢解顺序由取代苄基芳环上的电子效应和底物对金属钯表面的亲和力控制。苄基的芳环连有供电子基时,其通常较易被脱除;反之,则较为稳定。然而芳环取代基的存在可能诱发不利的空间效应并影响键合所需的平面几何结构,从而降低氢解活性[56]。
Sajiki 等[57−58]发现游离胺可能会抑制脂肪族/芳香族苄基醚的氢解,并考察了邻位取代的吡啶衍生物对脂肪族苄基醚加氢脱保护的抑制作用,发现吡啶环上的氮孤对电子周围的空间位阻可以减弱该含氮碱与金属钯的相互作用(其对钯表面的亲和力和对钯表面活性位点的抑制作用),从而可能避免Pd/C 催化剂的失活;其次,含氮碱的两氮原子之间的C—C 链长度在抑制芳香族苄基醚的氢解反应中起至关重要的作用。Yin 等[59]在优化雷贝巴坦(relebactam)关键羟胺中间体的合成路线时发现三乙胺和DABCO 可显著提高苄基醚的氢解速率,而2,6−二叔丁基吡啶对该反应具有抑制作用,这与上述Sajiki等的说法不一致。
此外,多数用于苄基醚氢解的催化剂也可能导致苄基芳环的饱和。Crawford 等[60]在氢解苄基醚和萘甲基醚期间发现样品中含有部分饱和醚,这无疑增大了产物的提纯难度。Ochocinska 等[61]以10%Pd/C 为催化剂,DMF−HCl 水溶液为溶剂,成功解决了萘甲氧基糖苷的氢解选择性问题,而无芳环饱和或酸不稳定基团分解等副反应。
连续流微反应器凭借其高传质和传热效率以及显著较短的反应时间被广泛用于脱保护反应条件的快速筛选和优化以及氢解产物的高通量合成。
Knudsen 等[62]利用H−Cube 反应器仔细研究了反应温度、底物流量和浓度对Boc−O−Bn−L−酪氨酸氢解的影响,发现增加底物的流量与浓度可加速催化剂失活,且温度的上升对维持催化剂活性有显著影响。在60℃的反应条件下,经过连续7 次加氢脱保护,底物的转化率仍接近100%(图2)。

图2 N−Boc−O−苄基−酪氨酸的连续氢解[62]Fig.2 Continuous hydrogenolysis of O−benzyl protected N−Boc−tyrosine[62]
Ekholm 等[29]在连续流加氢微反应器中以Pd/C为催化剂,MeOH−EtOAc 为混合溶剂,并通过高压柱塞泵将1 mg/ml的(3R,4R,6S)−3,4,5−三(苄氧基)−2−[(苄氧基)甲基]−6−甲氧基氧烷溶液以1 ml/min的流量送入系统进行苄基醚脱保护处理,产物甲基−α−D−吡喃甘露糖苷的收率高达95%(图3)。

图3 甲基−α−D−吡喃甘露糖苷的连续合成[29]Fig.3 Continuous synthesis of methyl−α−D−mannopyranoside[29](1bar=0.1 MPa)
Desai 等[63]在H−Cube 反应器中填充10% Pd/C,对含有酰胺键、碳碳双键的DHPM 苄基酯1a~1d 进行苄酯脱保护以获得相应的DHPM 酸(表2),为后续3,4−二氢嘧啶−2−酮衍生物(可用于制备抗高血压药和钙拮抗剂)的高通量合成奠定了基础(图4)。

图4 DHPM苄基酯的连续氢解[63]Fig.4 Continuous hydrogenolysis of DHPM benzyl esters[63]

表2 连续流条件下四种DHPM 苄基酯的氢解Table 2 Hydrogenolysis of four DHPM benzyl esters under continuous flow conditions
Zhang等[64]使用H−Cube反应器对四氢萘酰胺衍生物(表3 a~d)和二氢茚酰胺衍生物(表3 e~g)进行苄基醚脱保护(图5~图6)。当氢气压力为0.1 MPa、反应温度为45℃时,前者的收率可达86%以上,后者在2 MPa 的氢气压力下也可获得86%以上的高收率。

图5 四氢萘酰胺类衍生物的连续氢解[64]Fig.5 Continuous hydrogenolysis of tetrahydronaphthalene amide derivatives[64]

图6 二氢茚酰胺类衍生物的连续氢解[64]Fig.6 Continuous hydrogenolysis of dihydroindene amide derivatives[64]
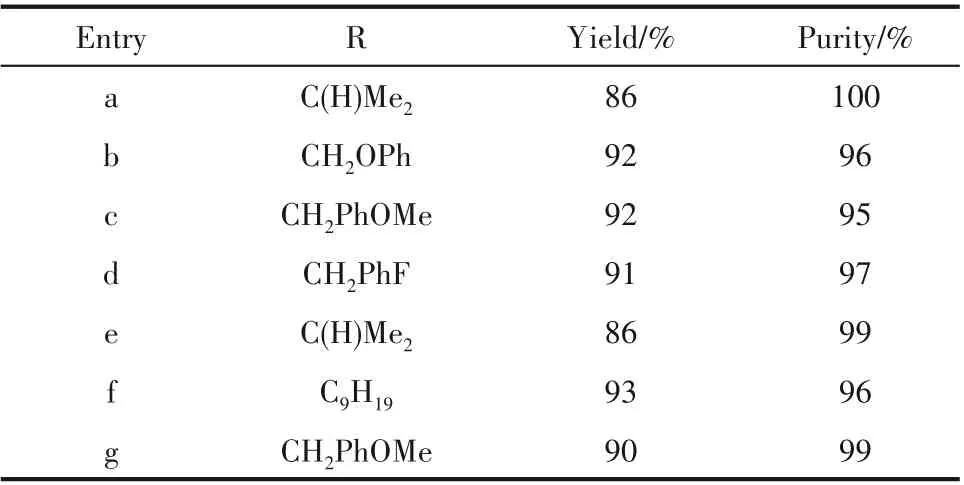
表3 双环羟基酰胺的合成Table 3 Synthesis of bicyclic hydroxy amides
2.1.2 N−苄基脱保护 苄基容易引入,但较难脱除,特别是N−苄基的脱保护[65−66]。相比于O−苄基加氢脱保护,其氢解过程通常需要更高的催化剂负载量、反应温度和压力[67]。受N−苄基保护的叔胺和季胺一般在室温与常压下较易被氢解,而伯胺或仲胺需要更高的压力(>4 bar)和温度(>40℃)[68]。同时N−苄基氢解产生的游离胺会与催化剂的活性中心产生强相互作用,从而削弱催化剂的部分活性甚至使其完全失效[42]。此外,Tanielyan 等[69]意外发现低压氢气有利于提高含吡啶酮和芳香氟化物结构的复杂苄胺的N−苄基脱保护速率,即提高产物的选择性。一种可能性是使用低压氢气可减小催化剂表面的氢气覆盖率,从而提高苄基在催化剂表面的吸附程度,加快脱苄基反应。
Jones 等[70]在H−Cube 连续流反应器中填充10%Pd/C催化剂,并以EtOAc/EtOH(1∶1)为混合溶剂,对N−苄基−2−苯乙胺进行氢解,实现了100%的转化率和89%的收率(图7)。该连续流反应体系有效解决了N−苄基脱保护问题,并为Boc 和Fmoc 保护基的酸解提供了有效替代方案。

图7 N−苄基−2−苯乙胺的连续氢解[70]Fig.7 Continuous hydrogenolysis of N−benzyl−2−phenylethylamine[70]
Darvas 等[71]使用实验室规模的连续流加氢装置对N−苄基−2−苯乙胺的脱保护也进行相关研究。其以EtOAc/EtOH 为溶剂,钯黑为催化剂,当底物浓度为0.05 mol/L 时,苯乙胺收率将近99.9%。相比于Jones等[70]的实验结果,其收率有显著提高(图8)。

图8 N−苄基−2−苯乙胺的连续氢解[71]Fig.8 Continuous hydrogenolysis of N−benzyl−2−phenylethylamine[71]
Baxendale 等[72]使用HEL 连续流加氢系统,并耦合微波和连续流技术,进行1−苄基−1,4,5,6−四氢环戊并[b]吡咯−2−羧酸乙酯的加氢脱保护,同时饱和其芳环生成相应吡咯烷作为雷米普利前体,其产物收率可达97%(图9)。
本课题组[33]采用微填充床反应器成功实现了N−二苯甲基的连续高效氢解脱除。该系统经过190 h 的连续运行,产物收率仍可维持在99.5%以上。此外,对比了传统间歇式反应器(250 ml)和微填充床反应器在相同反应温度和压力下的反应性能,发现后者单位体积单位时间的原料转化量是前者的100 倍,这极大地显示了连续流微反应器的氢解优势。

图9 1−苄基−1,4,5,6−四氢环戊并[b]吡咯−2−羧酸乙酯的连续氢解[72]Fig.9 Continuous hydrogenolysis of ethyl 1−benzyl−1,4,5,6−tetrahydrocyclopenta[b]pyrrole−2−carboxylate[72]
2.2 脱苄氧羰基反应
苄氧羰基常用于保护胺类物质,一般出现于多肽的合成过程中。相比于脱苄基,其氢解产物还包括CO2。Müslehiddinoğlu 等[73]在间歇加氢反应釜中以Pd/C 为催化剂,对N−苄氧羰基的氢解过程进行了研究。结果表明,CO2对催化剂的影响较大,其会改变脱保护反应的速率与级数。此外,他们分别利用碳酸钾和三乙胺进行CO2的吸收与溶液pH 的调节,发现两种方式均可降低催化剂的中毒程度,同时借助Langmuir−Hinshelwood 机理描述苄氧羰基的氢解过程,提出产物中间体的形成步骤为决速步骤。而戴云生等[42]表示N−苄氧羰基型保护基的氢解体系不局限于碱性环境,也可为中性或酸性,后者可能是利用有机胺的质子化,从而降低催化剂的中毒程度。黄建珍[28]利用填充床振荡流反应器合成氨曲南主环,并借助装置的平推流与无梯度特性将生成的CO2迅速带走,使氢气分压保持恒定以探究反应的宏观动力学。其同样认为产物中间体的形成步骤为决速步骤。此外,Gaunt等[74]发现富电子的2−萘甲基氨基甲酸酯的氢解活性远不及苄氧羰基,这与此前该团队研究的2−萘甲基−苄基体系形成鲜明对比。其推测这可能与N−苄氧羰基的自身反应性较强有关。
Oyamada 等[35]将高活性、低浸出率、无溶胀的Pd/(PSi−Al2O3)催化剂运用于连续流动反应器中,并可能考虑到Cbz−L−丝氨酸易溶于醇溶剂,产物丝氨酸易溶于水,而选择乙醇/水(1∶4)为混合溶剂使整个反应在溶液中进行,最终成功实现Cbz−L−丝氨酸的加氢脱保护[图10(a)]。此外,该团队发现Pd/(PSi−Al2O3)催化剂在此氢解系统中可至少维持8 h的高活性,且在反应期间无金属钯浸出。

图10 N−苄氧羰基的连续氢解[35,70−71,75,78−79]Fig.10 Continuous hydrogenolysis of N−benzyloxycarbonyl[35,70−71,75,78−79]
Jones 等[70]在H−Cube 反应器中填充10%Pd/C 催化剂,并在常压、50℃的反应条件下,对含双键结构的(2−(1H−吲哚−3−基)乙基)氨基甲酸苄酯进行N−苄氧羰基脱保护,最终获得较高收率的色胺[图10(b)]。Darvas 等[71]利用实验室规模的连续流加氢装置对0.05 mol/L 的上述底物溶液也展开了相应脱保护研究,相比于Jones 等的实验结果,其产物收率有显著提高,可达95%[图10(c)]。
苯乙胺是制备药物或染料的重要中间体,主要用于合成抗抑郁药、支气管扩张药等。Kobayashi等[75−77]利用聚合物包埋法将钯络合物固载于玻璃微通道内壁,同时要求气−液两相以环流形式与催化剂相接触,在1 min 内成功实现苯乙基氨基甲酸苄酯的N−Cbz 脱保护,且产物收率接近100%[图10(d)]。
Ladlow 等[78]利用H−Cube 反应器依次对预先合成的Cbz−L−Val−L−Phe−OMe 等肽链进行连续流氢解[图10(e)]。为解决较长肽链的溶解性问题,该团队选择二甲基甲酰胺为氢解溶剂。实验结果表明,受苄氧羰基保护的较长肽链在60℃和0.1 MPa 下均可获得不同程度的脱保护,这为将来的药物和多肽合成提供了新的可能。
Clapham 等[79]设计、构建了一套自动化连续流加氢装置以快速筛选与优化2−[(4−苯氧基苯基)氨基甲酰基]吡咯烷−1−羧酸苄酯的脱保护反应条件,并借助液质联用分别对30、40、60 和80℃反应温度下的样品进行检测,发现在60℃、0.1 MPa 的反应条件下该氢解反应可得到100%的转化率[图10(f)]。
3 结论与展望
连续微反应加氢技术是一种原子经济且高效绿色化的合成手段。相较于高压间歇加氢釜等传统反应器,连续流微反应器可利用其微通道优势解决气−液−固三相界面接触面积小、相间传递速率低等问题,其高效的气液传质效率与平推流特性还可实现高选择性脱保护,并显著缩短反应时间。此外,加氢脱保护通常需在一定压力下进行,以增加氢气在溶剂中的溶解度及其在催化剂表面的吸附程度,故存在一定操作危险性,而连续微反应器凭借其体积小、持液量小、氢气滞留量低等优势可实现安全可控的生产目的。催化剂、溶剂和添加剂的合理选择也可进一步提高氢解产率。Pd/C、Pd(OH)2/C 或两者的混合物通常被视为高活性脱保护催化剂,且因固载化而无需分离,操作安全性较高;低级醇、四氢呋喃、乙酸乙酯、乙酸和二甲基甲酰胺等极性溶剂为常见的脱保护溶剂,而甲醇和乙醇因低毒性、低成本和高溶解性,通常被视为最佳溶剂;有机酸或含氮碱的引入可选择性地促进或抑制苄基,从而提高产物选择性;双相混合溶剂的应用还可助于氢解产物与保护基的相分离,既能有效解决目标产物的分离问题,又能避免有机组分快速固化而带来的管线堵塞问题。目前连续微反应加氢技术在脱保护反应中的报道大多基于实验室规模的合成与研究,本课题组已经成功实现了连续流加氢微反应器的放大,并运用于脱苄基产品的吨级生产。该成果表明,连续微反应加氢技术具有过程安全性高、反应时间短和催化剂成本低等特点,相比传统的釜式加氢具有明显优势。但同时,该技术还面临过程易堵塞、工艺开发时间长等问题。相信随着该技术的不断进步,其在未来将被广泛运用于医药和精细化工等诸多领域。
