表面诱导药物多晶型成核的研究进展
2021-03-06林家伟石鹏龚俊波吴送姑
林家伟,石鹏,龚俊波,吴送姑
(天津大学化工学院,化学工程联合国家重点实验室,天津300072)
引 言
多晶型是指同种分子由于组装方式不同而形成的两种或两种以上的不同的结构,主要包括构型多晶型和构象多晶型[1−2]。在制药领域,药物的多晶型十分常见。 同一药物活性组分(active pharmaceutical ingredient,API)不同的晶型往往会表现出不同的物理化学性质,如晶习、溶解度、溶出速率、熔点、颜色、硬度、密度等,这些性质又与后续的压片性能、药物的稳定性以及生物利用度具有很大的关系,所以制备和控制API 的晶型十分必要[3−7]。控制晶型的关键步骤是成核过程,晶核中分子的组装方式决定了产品的晶型,所以成核过程的控制是关键。通过控制成核过程的条件,可使溶质分子以不同的方式进行组装,从而得到不同的晶型。传统晶型的调控方法主要是基于结晶条件的控制,包括溶剂、温度、过饱和度、晶种[8]、添加剂[9−12]、pH[13−18]等。近些年来,随着研究的进一步深入,涌现出许多新型的晶型调控策略,主要包括离子液体[19−22]、微通道技术[23−24]、表面诱导[25−26]以及激光诱导[27]等。表面诱导策略主要是指在异相成核过程中通过引入具有特殊物理或化学性质的异质表面,在不改变溶剂、温度等条件下,利用异质表面与溶质分子的相互作用干扰分子组装达到晶型调控的目的。晶体、无定形甚至流体与乳泡的界面都可以作为异质表面,而且异质表面的材料可以是有机材料也可以是无机材料[28]。近些年来,表面诱导成核法在药物新晶型的发现以及对亚稳晶型的调控方面得到了广泛地研究。本文介绍了异质表面影响多晶型成核的机理——化学作用和几何作用,综述了用于诱导药物多晶型成核的四类异质表面——聚合物、自组装单层膜(SAMs)、有机小分子晶体和凝胶,并总结了异质表面选择与设计的常用策略,相关研究概况示意图如图1所示。
1 异质表面影响多晶型成核机理
1.1 化学作用
异质表面与溶质分子特定的化学作用对于成核以及晶型的选择具有重要的作用。在无表面辅助诱导的溶液结晶过程中,只有溶质−溶质、溶剂−溶剂、溶质−溶剂之间的相互作用,异质表面的引入使得表面−溶质、表面−溶剂之间的相互作用力参与到成核的过程中。有利的异质表面基团与溶质分子相互作用可以局部增大溶质分子浓度促进溶质分子的有序排列,这种有利相互作用主要包括非特异性吸附、氢键、π−π 作用以及其他弱的分子间作用力。在非特异性吸附的情况下,表面通过降低成核所需的表面自由能来促进成核[26]。如果表面与溶质分子具有更强的相互作用,例如强氢键、π−π 作用,那么异质表面便更可能会干扰分子的组装方式。表面诱导成核中表面化学作用的一个关键点是晶体的不同晶面暴露的官能团不同,从而与异质表面产生特定的分子间作用力,进而影响溶质分子组装,导致晶型的选择性成核[28]。此外,化学作用还会影响晶体−溶液、晶体−表面、溶液−表面之间界面能的平衡状态,从而影响多晶型异相成核的结果[29]。
1.2 几何作用
异质表面的几何作用对多晶型的成核同样具有选择作用。对于晶体材料,几何作用主要是指当晶体基底与API晶格参数类似或者当晶体的晶面夹角与成核前聚体相匹配时,成核晶体通过与模板晶体的覆盖面进行外延生长,这种几何作用称之为外延作用[30−32]。对于非晶体表面,表面形貌对于晶型的诱导具有不可忽视的作用,例如非晶体表面纳米孔隙的大小、形状等都对溶质分子的组装具有约束作用,从而对成核过程产生影响[33−35]。
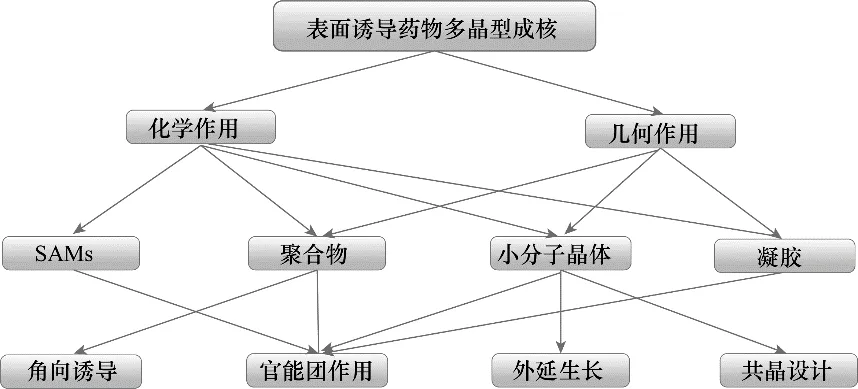
图1 表面诱导药物多晶型成核的研究概况示意图Fig.1 Schematic diagram of overview of surface−induced nucleation of drug for controlling polymorphism
1.2.1 外延作用 外延作用是一种二维形式的非均相成核,其基底是材质上不同或者晶体形态不同的材料,当基底与沉积层的晶体学取向发生匹配时,外延晶体表现出特定的取向。人们对外延的兴趣主要集中在硅基板上沉积半导体的光电应用,或者利用固体/蒸气过程以及均匀缓冲层来制造纳米器件。对于有机晶体,利用外延作用控制晶型已有诸多的报道,外延作用可以理解为异质表面与溶质分子的晶体结构具有相似性,这种相似的结构可以有效降低成核能垒,有利于表面选择该特定晶型的成核与生长。外延作用主要分为二维外延作用与台阶外延(ledge directed epitaxy, LDE)作用[30−31]。如图2 所示,二维外延作用需要异质表面与其接触的晶面能够进行晶格匹配,由于晶格匹配作用,可使成核分子以特定的方式组装并在异质表面成核[30]。利用晶体外延的几何空间分析程序(GRACE)通过几何晶格匹配计算可预测溶质晶体结构与模板之间的外延关系[32]。Ward 等[31]提出了LDE 的作用机制,当两个紧密排列的表面之间的二面角对应于某个晶型晶体的一个内角时,该晶型可以选择性成核。相对于二维外延作用,目前关于LDE 作用机制的文献报道较少。
1.2.2 表面形貌 缺陷、位错和不规则的结构可以作为异质成核位点,促进分子的有序排列,因此异质表面诱导成核的能力与它的形貌密切相关[26]。异质表面孔道的尺寸、形状和粗糙度等对成核和晶型的选择具有重要的影响,特别是表面孔道。关于表面孔道的影响,可以通过聚合物薄膜或者玻璃表面制备成不同大小、角度的孔道,通过“空间限制效应”研究对晶型的选择性[36−39]。研究表明,当在纳米孔径内发生成核时,如果溶质临界晶核大小与孔径大小相对应,那么可通过“填孔”机制提高成核速率[40];在尺寸接近临界核的孔隙中,可以通过控制孔隙的大小来调控晶型,这主要是由于不同的晶型临界成核尺寸不同,当孔隙的尺寸小于某一晶型的临界成核尺寸时,该种晶型的成核会受到抑制[41]。此外,可以利用纳米孔隙的限制使亚稳态晶体稳定存在,并阻止其向稳定晶型转化[42]。孔隙的形状对于成核与晶型的选择也不可忽略,尤其是当孔隙的形状与晶体晶面的内角相匹配时[43]。表面粗糙度显著影响溶剂在表面的润湿行为,影响溶质分子与表面的作用机会,从而影响异质表面对于晶型的调控[39]。
事实上,在很多情况下化学作用与几何作用对表面诱导成核的影响是互补的,甚至表面化学占据更重要的作用。Trout 等[38]在以具有纳米压印的聚合物薄膜作为基底时发现聚合物薄膜与溶质分子的相互作用是诱导作用的前提,在此基础上孔道的结构会进一步促进成核。Ward 等[32]研究了几种有机晶体在晶体表面上成核过程中的化学作用和外延作用,研究表明与外延作用相比,化学作用具有明显的优势。总地来说,晶体在异质表面上的成核和生长仍然是一个复杂的课题,在许多情况下,化学和几何相互作用的微妙平衡决定了结晶的结果。
2 异质表面的种类
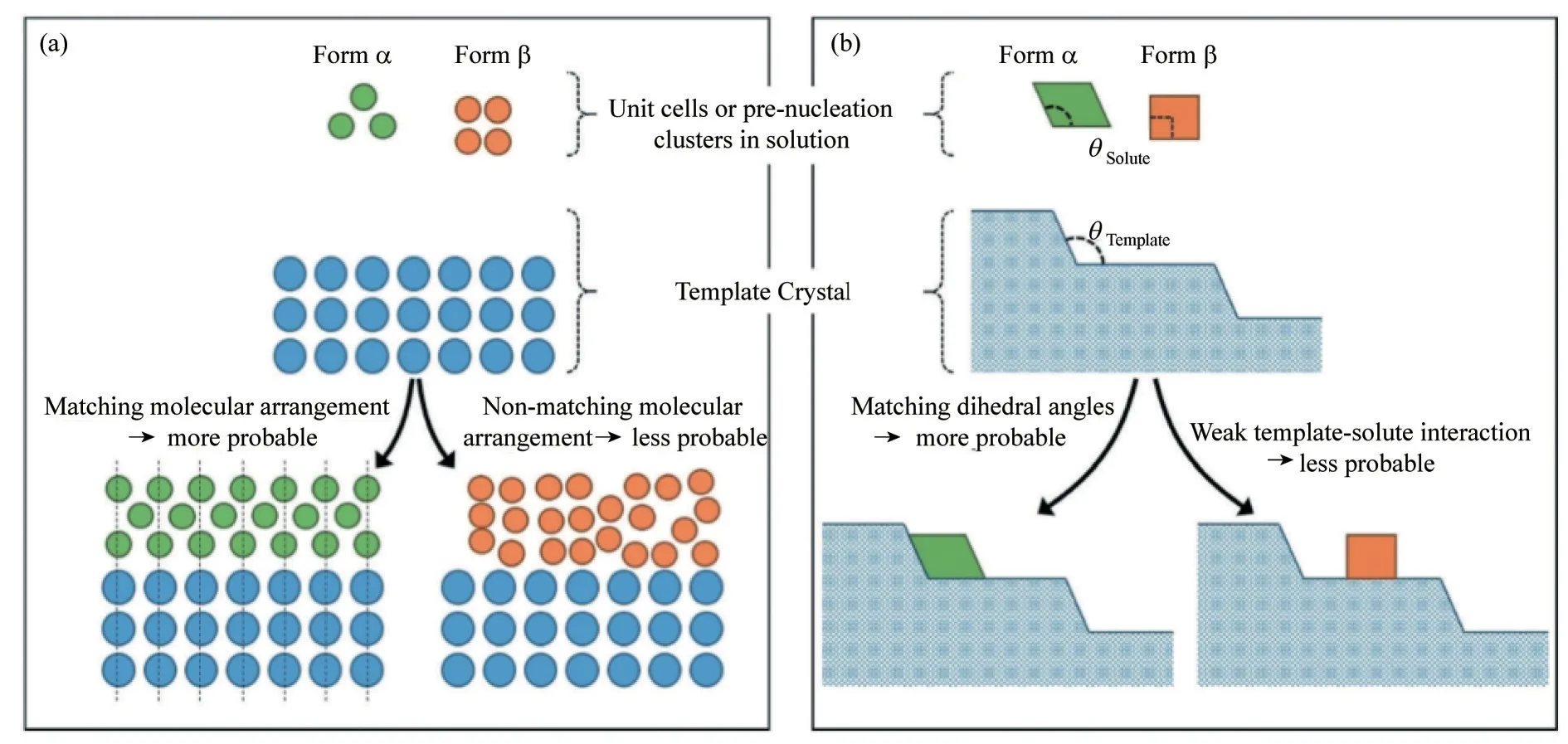
图2 二维外延(a)与LDE成核(b)示意图[28]Fig.2 Schematic diagram of two−dimensional epitaxial nucleation(a)and LDE nucleation(b)[28]
异质表面是指在溶质分子成核过程中引入的具有特殊物理化学性质的且不溶于所处溶剂体系下的表面基底,由于异质表面特殊的物理化学性质,可干扰溶质的分子组装,从而有效降低成核诱导期并对多晶型进行调控。近年来,基于化学作用、几何作用,表面诱导作用在药物新晶型的发现以及对亚稳晶型的调控方面得到广泛关注。常用的异质表面主要包括聚合物、自组装单层膜、有机小分子晶体以及凝胶等。因其易于对表面的化学结构进行改性,以及新方法实现了对其表面几何结构的可控性调整,聚合物成为了研究者们应用最广泛的一类材料。自组装单层膜在利用化学作用调控晶型方面具有显著的优势,其表面可赋予致密且丰富的官能团,并且制备方法较为简单。与聚合物和自组装单层膜相比,小分子晶体结构的长程有序性以及结构的简单性使之成为利用外延作用调控成核的优先选择。凝胶相结晶作为一种新兴的方法在异相成核方面展现了巨大的潜力,其几何结构的可微调性以及丰富的化学结构为溶质分子提供了一个特殊的受限环境,使之成为可媲美聚合物的一种新的异质表面。此外文献[43]也报道了利用硅酸盐等无机材料作为表面成核基底,但其对晶型的诱导作用并不明显。
2.1 聚合物
聚合物是表面诱导成核的最常见的一种异质表面[44],近些年来基于高通量的筛选方法,利用聚合物诱导成核发现了大量的药物新晶型。Roy 等[45]采用熔融结晶的方式,以4−(苄氧基)苯腈改性的聚苯乙烯作为异质表面,成功发现了苯巴比妥的一种新晶型X;López−Mejías 等[46]使用非极性芳香类聚合物成功筛选出托灭酸3 种新的亚稳晶型,随后又利用不同的聚合物薄膜作为异质表面,分离得到氟灭酸6 种新的晶型,从而使之具有8 种晶型,创造了当时具有已知晶体结构晶型数目最多的药物活性分子的新纪录。
利用聚合物作为异质表面必须保证在所处溶剂体系中不溶,将丙烯酸酯等紫外光敏单体交联合成高分子膜是研究者们常用的一种方法。此外还有基于对聚合物溶液处理的方法,包括溶剂蒸发、旋涂法、蒸汽诱导相分离等都可用于制备相应的聚合物薄膜[26]。一旦聚合物薄膜在促进成核的有效性得到证实,其更复杂的表面形貌将被进一步探索,研究者们利用各种技术在聚合物薄膜表面赋予其纳米尺度的图案,利用图案的几何效应对溶质分子的成核进行调控。事实上,无论是光滑的聚合物薄膜还是具有特殊纳米形貌的聚合物表面,其选择合成的单体大多具有特殊的官能团,使之合成的聚合物侧链官能团易与溶质分子相互作用。这主要是由于所使用的聚合物多为无定形,没有明显规律的分子堆积方式,外延生长并不能作为聚合物诱导成核的一种方式,其作用机制更多的是基于界面的化学作用,在此基础上,表面形貌的作用会进一步促进溶质分子的组装。
López−Mejías 等[47]在聚合物诱导药物成核多晶型方面展开了大量前瞻性的研究,他们认为聚合物诱导多晶型成核的主要作用机制是溶质分子通过界面的相互作用在聚合物表面定向排列,这种不同的界面作用主要源于不同的聚合物暴露不同的侧链官能团,从而导致晶面沿择优取向面成核。Song等[48]最近的研究发现溶质分子与聚合物表面的界面作用还受溶剂效应的影响,这主要是由于不同的溶剂对于聚合物薄膜的润湿行为不同。他们发现以glass(玻璃)、PTFE(聚四氟乙烯)、PET(聚对苯二甲酸二醇酯)、POM(聚甲醛)作为异质表面在丙酮、1,4二氧六环、乙腈溶液中获得的是吡嗪酰胺的δ 晶型或者α 与δ 晶型的混合物,但以PA66(尼龙−66)作为表面在丙酮和乙腈溶液中获得了纯的α晶型。他们测量了溶剂与聚合物表面之间的接触角并模拟了在不同溶液中PA66表面与溶质分子的相互作用,发现在乙腈与丙酮溶液中PA66 表面容易被润湿,且在这两种溶液中吡嗪酰胺可与PA66 形成较强的氢键,这有效促进了α晶型的出现。
事实上聚合物表面和溶质介质在晶型选择上是可以相互独立作用的,因为在没有溶剂的情况下以及在不同溶剂的存在下,都可以根据它们提供氢键的能力改变晶型。McKellar 等[49]使用无溶剂熔融冷却结晶的方法,使用一系列聚合物对吲哚美辛的晶型调控进行了研究,实验结果表明具有不同官能团的聚合物诱导效应不同,具有苯基和四氢呋喃侧链的聚合物表面更容易诱导产生α晶型。
目前,聚合物诱导成核多是基于界面的化学作用,聚合物的选择需要进行大量的试错。未来通过阐明在不同的结晶条件下每个优先成核面发生的不同的分子间相互作用,将实验与计算模拟相结合,可提高聚合物筛选的效率;并且可以进一步促进对聚合物诱导多晶型成核的认识。另外通过对结晶过程中多晶型选择性成核的分子间相互作用的进一步明确,将有助于进一步发展利用聚合物异质表面控制和选择药物晶型的更好方法。
2.2 自组装单层膜
自 组 装 单 层 膜(self−assembled monolayers,SAMs)是指有机功能化的分子通过强键作用力(如静电作用力、共价键、配位键等)自发地吸附在固/液或气/固界面,与界面形成的高度有序的、低缺陷的单分子层[50]。如图3 所示,自组装膜主要由极性头基、碳链骨架、末端基团三个部分组成,极性头基通过强键作用力吸附在基底表面(主要包括过渡金属、金属氧化物、玻璃、云母等),末端基团通常是特殊设计的官能团分子以便与溶质分子相互作用。根据极性头基与基底表面材料的不同,自组装单层膜主要分为脂肪酸自组装、有机硫自组装、有机硅烷自组装。目前关于自组装膜的研究与应用70%以上集中在有机硫,特别是Au/硫醇自组装,这主要是由于金的表面具有较好的稳定性,Au−S具有较高的连接强度,并且其制备过程易于控制。
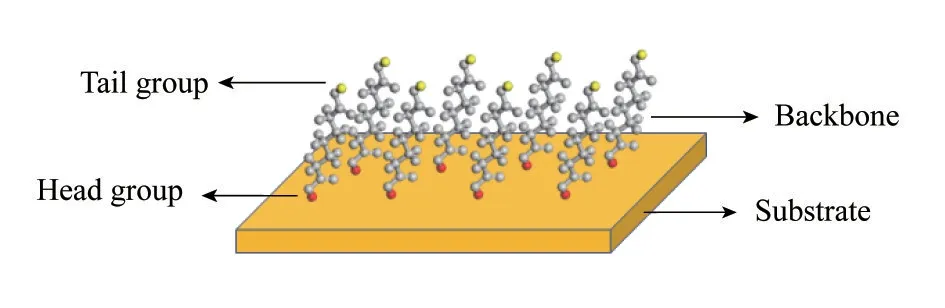
图3 自组装单层膜结构示意图Fig.3 Schematic diagram of SAMs structure
由于SAMs 具有的高度有序的分子组装表面,被广泛用于调控晶体的成核与生长过程,包括晶核密度、晶体大小、晶面取向、形态、晶型、稳定性等[51]。自组装膜调控多晶型主要是通过其表面基团与晶体分子之间产生的界面的化学相互作用,包括氢键作用力、π−π 作用、空间几何作用以及其他的弱相互作用。
氢键是SAMs 调控药物多晶型的一种重要的化学作用力,是形成药物稳定结构以及晶型调控的一种重要手段。Cox 等[52]研究了含有不同表面官能团的SAMs 对于茶碱的影响。实验发现,在含有羟基或者羧基官能团的SAMs 表面,茶碱以无水晶型存在,这主要是由于羟基或者羧基能够提供氢键供体,可与其(200)晶面暴露的羰基作用,从而降低成核能垒。
π−π作用是指具有π电子体系的芳环之间产生的由非共价键连接的一种分子间作用力,在SAMs调控药物多晶型方面得到广泛的应用。Quist 等[53]利用末端基团为联苯的SAMs 对抗惊厥药物卡马西平晶型的选择作用进行了研究。他们认为卡马西平分子二聚体与SAMs 末端的联苯形成π−π 作用,这种π−π 作用有利于卡马西平分子以特定形式进行分子组装,从而有利于产生亚稳的晶型Ⅱ。Yang等[54]研究了不同表面功能化的SAMs 对甲灭酸多晶型的影响。亚稳晶型Ⅱ易于在吡啶或羧基表面功能化的SAMs 表面结晶,这主要是由于具有吡啶或羧基的SAMs 可与甲灭酸形成π−π、C—H…π 作用,减小了甲灭酸的极性,降低了成核能垒,从而促进了亚稳晶型Ⅱ的成核。
SAMs 界面与溶质分子的其他弱相互作用,也被报道用于对晶型的调控。Hiremath 等[55]利用SAMs 界面与2−碘−4−硝基苯胺之间所形成的I…NO2作用,成功制备出2−碘−4−硝基苯胺的亚稳晶型。Cox 等[56]利用全氟羟基为末端基团的SAMs 的“不沾性”阻碍了吲哚美辛亚稳晶型的非均相成核,利用“Ostwald 熟化”以亚稳态α 晶型的消耗为代价促进稳定晶型的生长,从而得到了稳定的γ 晶型。Bolla等[57]利用SAMs与有机金属框架(MOFs)制备了一种新的表面金属有机框架基底(SURMOF),由于弱的超分子作用力,扑热息痛被吸附在MOF 孔道里,诱导了扑热息痛晶型Ⅱ的成核与生长。
2.3 有机小分子晶体
有机小分子晶体作为异相成核的模板表面对药物分子多晶型进行调控的主要作用机制是基于溶质分子在有机小分子特定晶面的外延匹配作用以及官能团的相互作用。实际上,目前利用有机小分子晶体作为异质表面的报道相对较少,主要是由于溶剂体系与小分子晶体的选择难以平衡,既要筛选到合适结构的小分子晶体,同时还须确保其在选择的溶剂体系下保持不溶,这就限制了有机小分子晶体的可选性。此外,结晶得到的晶体与有机小分子晶体的分离也是潜在的问题,目前所采取的策略是在添加量少的基础上不对两者的混合物进行分离。所以相比于聚合物与SAMs,有机小分子晶体的筛选具有一定的难度。相关文献报道[30,58],通过培养形状、大小较好的单晶以其作为模板表面,采用升华结晶的方式可以有效规避上述存在的问题,同时可探究外延作用对于多晶型选择的影响。
尽管如此,有机小分子晶体也有其特殊的优势,晶体长程有序的结构是聚合物与SAMs 所不具备的,这更有利于晶型的诱导作用。Zhang等[59]研究了晶体结构对于多晶型成核的影响,分别以呈晶态和无定形态的氢氯噻嗪为模板表面探究了对吡嗪酰胺(PZA)晶型的调控作用。实验发现呈现晶态的氢氯噻嗪能够诱导产生γ 晶型的PZA,而以无定形状态的氢氯噻嗪作为模板表面产生的是α 晶型的PZA。他们认为,晶体模板表面的官能团较为有序,具有一定的取向效应,这种取向效应可以有规律地与溶质分子相互作用,从而诱导产生γ 晶型。无定形分子表面官能团的无序排列,虽然可以吸引溶质分子聚集在表面周围,但与溶质分子相互作用的官能团并不是长程有序的,所以并不能干扰PZA 的分子组装方式,如图4所示。
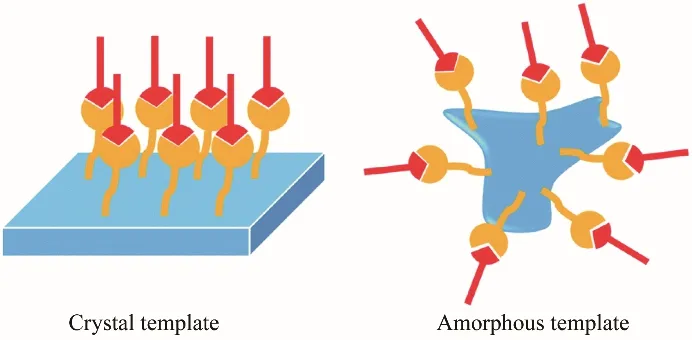
图4 晶体和非晶态模板的溶质分子排列示意图[59]Fig.4 Diagram of solute molecule arrangements with crystal and amorphous templates[59]
此外,有机小分子晶体异质表面并不局限于与API分子不同的物质。晶种法作为一种常规的晶体制备策略,已经被广泛应用到实验和生产当中。当使用稳定晶型的晶体作为晶种时,一般认为结晶过程最终产生的晶型与晶种相同,而有研究发现当以稳定晶型的晶体作为晶种时也会得到亚稳晶型的晶体。Park 等[60]以晶种法大量制备多奈哌齐晶型F时,意外发现了一种新的亚稳晶型K。在不添加晶种时,过饱和度S>12 时,晶型K 可以通过均相成核形成;然而当以晶型F作为晶种时,在较低过饱和度(S=2~3)时,便可产生亚稳晶型K。对两种晶型的结构进行研究可以发现,这两种晶型存在一个紧密相关的晶面,对应于晶型F 的(010)面。当以晶型F 的单晶作为模板表面时,通常可以观察到K 型外延晶体生长在(010)面。这种诱导效应可理解为由于二维的晶格匹配作用,导致了较低的界面能,从而降低了成核能垒。
应当注意的是,不同于聚合物与自组装单层膜,有机小分子晶体各个晶面暴露的官能团不同,并且溶质分子在各个晶面外延生长的能力不同,所以小分子晶体的晶习调控就显得尤为重要,使用暴露有优先作用晶面的晶体,将有助于发挥小分子晶体的诱导作用。
2.4 凝胶
凝胶相结晶是异相成核的一种方法,它提供了一种既能控制表面几何结构又能控制表面化学结构的体系。凝胶中的小网格尺寸将吸附的溶液分割开来,限制被吸附的溶质分子的流动性,从而为结晶过程提供一个“受限环境”,凝胶剂−API 的化学作用则提供了空间约束下溶质分子组装方式的进一步调整[61−63]。虽然,部分凝胶属于聚合物的范畴,但由于其作用机制与聚合物表面并不同,聚合物表面大多是以薄膜或是粉末颗粒的方式引入溶液体系中,而凝胶则是通过非共价相互作用或者化学键自组装为一维纤维状长链,再由这些纤维状长链纠缠在一起,最终形成凝胶的骨架。通过表面张力将溶剂分子吸附在凝胶骨架中,就形成了凝胶[64]。
Diao等[61]使用微凝胶研究了对卡马西平和ROY成核多晶型的影响(图5)。通过调控交联聚乙二醇二丙烯酸酯微凝胶的尺寸,可选择性诱导晶型Ⅱ的卡马西平成核。此外,微凝胶使得对于ROY 的R 晶型选择性提高了20 倍。他们认为由微凝胶网络所施加的成核模板效应和空间约束可能是实现多晶型选择性的关键。由于交联的聚合物是无序的,二维表面外延的作用机制被排除,界面处的相互作用被认为是诱导成核的重要因素。
Foster 等[65]模仿ROY 的官能团设计合成了双尿素凝胶,在这种甲苯溶液的凝胶中,得到ROY 亚稳晶型R,而在其他形式的凝胶中ROY呈稳定晶型Y。构象与晶体结构预测的方法揭示了表面凝胶与ROY 的构象匹配可能是诱导产生亚稳晶型R 的原因。
Araya−Sibaja 等[66]利用金属配位的单宁酸超分子凝胶对咖啡因、卡马西平和吡罗昔康结晶过程的影响进行了研究。结果表明,凝胶相的引入对卡马西平与吡罗昔康晶型的成核有选择性。在凝胶相中,卡马西平二水合物的晶型占据主导地位,而对照组中主要以晶型Ⅲ的形式存在。对于吡罗昔康,晶型Ⅰ、晶型Ⅱ的混晶存在于凝胶相中,而在空白对照组中,吡罗昔康呈现纯的晶型Ⅰ。
Banerjee等[67]在二甲基亚砜溶液中,将氧化的纤维素纳米晶体与十八烷基胺混合制备凝胶,磺胺甲唑、磺胺吡啶和磺胺甲嘧啶在凝胶化步骤之前被添加到混合物中,使其在疏水区域中结晶(图6)。结果发现,得到的磺胺甲唑呈无定形,而磺胺吡啶以晶型Ⅰ、晶型Ⅲ以及晶型Ⅳ的混晶形式结晶,磺胺甲嘧啶以晶型Ⅰ和晶型Ⅱ的混晶形式结晶。
图5 凝胶网格尺寸对晶体成核的影响示意图[61]Fig.5 Schematic illustration of the effect of gel mesh size on crystal nucleation[61]
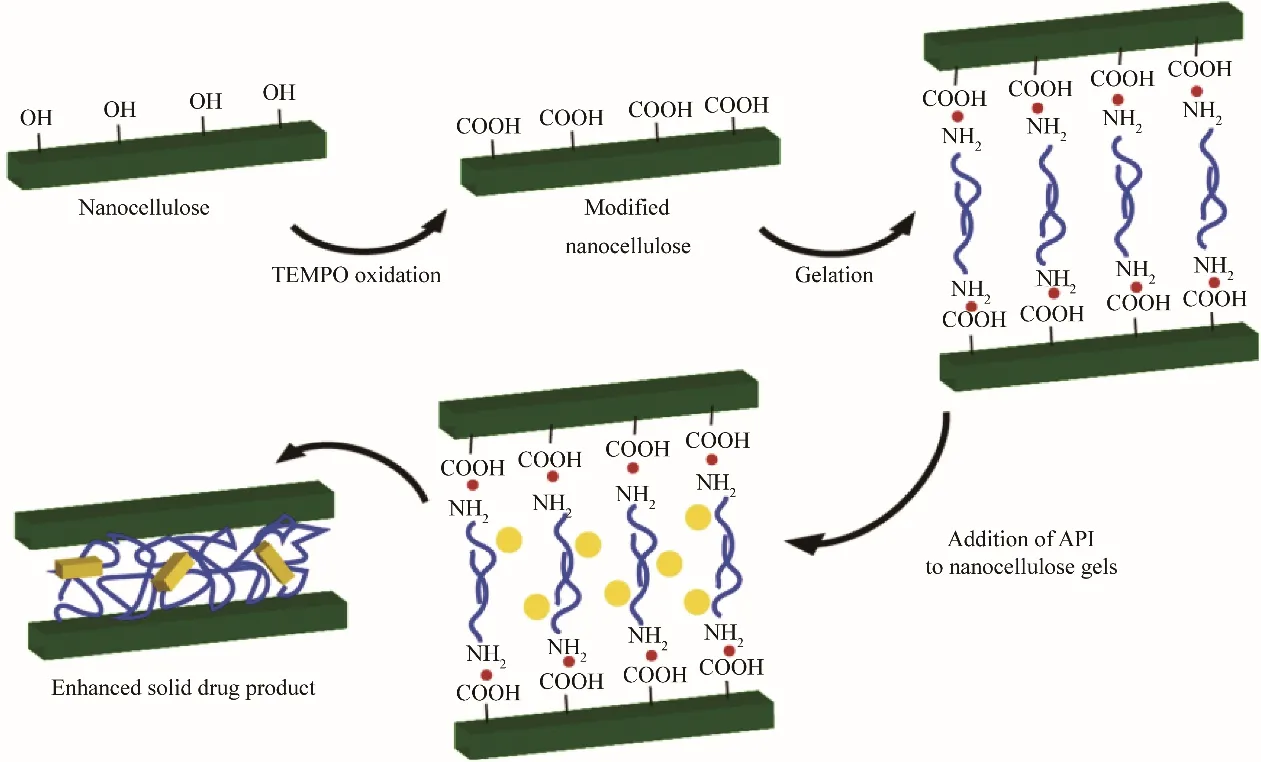
图6 用于药物结晶的表面改性纳米纤维素有机凝胶的示意图[67]Fig.6 Schematic representation of surface modified nanocellulose organogels used for pharmaceutical crystallization[67]
凝胶相结晶作为一种新兴的结晶方法,目前的研究方向主要集中在凝胶剂的设计为结晶提供特定的结晶环境与成核位点,但由于机理的不明确,目前对于晶型的调控还具有一定的盲区,因此相关报道较少。
3 表面选择与设计方法
目前由于表面诱导作用的机理并不明确,异质表面的设计和选择还处于盲筛的水平,需要经过大量的试错,如何更为高效地设计与选择合适的异质表面一直成为困扰研究者的难题。目前对于异质表面的选取更多是基于官能团的相互作用,但这种选择策略较为宽泛,对于异质表面的选择与设计并没有太强的针对性。近年来随着对异质表面诱导成核机理的进一步认识,一种基于化学作用的“共晶设计”的策略得到开发,此外利用几何作用“外延生长”、“角向诱导”的设计策略展现出了巨大的潜力。
3.1 基于官能团相互作用的表面设计
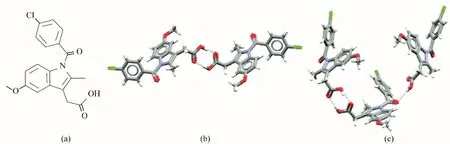
图7 吲哚美辛的分子结构(a),γ晶型的分子间作用与构象(b),α晶型的分子间作用与构象(c)[68]Fig.7 Chemical structure of indomethacin(a)and the intermolecular interactions and molecular conformations in the γ−form(b)and α−form(c)of indomethacin[68]
图8 6种功能化的晶体基底[68]Fig.8 Six kinds of functional crystalline substrates[68]
异质表面官能团可与溶质分子产生特定的相互作用,通过选用含有与溶质分子官能团类似或者易于发生分子间相互作用的官能团的异质表面,可达到干扰溶质分子组装的目的。尽管这种表面设计策略并不高效,但无论是选用聚合物、SAMs 还是小分子晶体作为异质表面,基于官能团相互作用的选取原则,一直是研究者们竞相选择的一种方式,同时也是其他设计方式的基础。
Wijethunga 等[68]基于官能团相互作用的原则,研究了一系列具有生物相容性的小分子晶体对吲哚美辛成核与多晶型选择的影响。如图7 和图8 所示,基于吲哚美辛含有的羧酸基团,他们选择了一系列具有—COOH、—NH2、—NH 等易于与之形成氢键官能团的小分子晶体作为模板表面,采用高通量的方法测量了不同表面对于吲哚美辛的成核诱导期。实验发现所有的模板表面均能有效缩短吲哚美辛的成核诱导期,且同一表面对不同晶型的诱导作用不同。基于拉曼光谱的晶面索引技术,识别了吲哚美辛与L−谷氨酸(L−glutamic acid)与L−组氨酸(L−histidine)的相互作用面,这些作用面暴露了较高密度的—COOH,揭示了溶质与表面官能团的相互匹配对于促进成核速率与外延生长的重要作用。
Bora 等[69]利用不同表面功能化的SAMs 对磺胺噻唑的晶型进行了调控,如图9 所示。磺胺噻唑是一种柔性药物,在结晶时会发生伴随多晶型的现象,在异丙醇溶液中以晶型Ⅱ、晶型Ⅲ、晶型Ⅳ的混晶形式出现。他们利用羧基、羟基、吡啶、苯并噻唑等六种功能化的SAMs 界面与其形成的包括氢键、C—H…π 以及π…π 的分子间作用力实现了对于磺胺噻唑伴随结晶的定向控制,其中在MSA(mercaptosuccinic acid)SAMs 表面形成了纯的晶型Ⅱ,而MAA(mercaptoacetic acid)SAMs表面生成了纯的晶型Ⅲ。 值得一提的是,在2MBT(2−mercaptobenzothiazole)表面由于C—H…π以及π…π的作用,发现了磺胺噻唑的一种新晶型。
3.2 基于共晶设计的表面
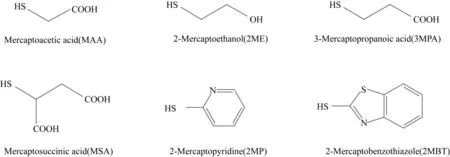
图9 用于制备SAMs的硫醇[69]Fig.9 Thiols used to prepare SAMs [69]
共晶设计的表面选择方式是在官能团相互作用的基础上进一步的延伸,实际上是基于“合成子”的设计理念。异质表面通常是可以与目标物质形成共晶的同类或者具有相同官能团的小分子晶体,这类异质表面的引入可以占据溶质分子堆积的特定位点,从而干扰分子的组装方式,进而达到对晶型进行调控的目的。共晶设计的表面设计理念来源于在共晶制备过程中偶然发现新晶型的现象,例如以烟酰胺和异烟酰胺作为戊氧苯硫脲配体的药药共晶制备过程中,同时发现了烟酰胺与异烟酰胺的新晶型,与稳定晶型相比,在堆积方式上都具有明显的区别[70]。
Zhang 等[71]基于共晶设计的表面筛选原则研究了磺胺类药物对吡嗪酰胺(PZA)的晶型调控作用。如图10 所示,PZA 在溶液中常常表现出二聚体(α、β、δ 晶型)的形式,链状的γ 晶型很难在溶液中得到。他们利用剑桥数据库(CSD)检索了可以与之形成共晶的配体,如图11 所示,在氢氯噻嗪与PZA 形成的共晶中,由于羰基与磺酰胺基团之间形成氢键从而使PZA 以链状的形式组装。基于此,选用了氢氯噻嗪、磺胺嘧啶等7 种磺胺类药物作为模板表面对PZA 的多晶型进行调控。实验结果与预期相符,吡嗪酰胺分子的羰基和磺胺模板分子的磺胺基之间形成氢键,这些优先的分子间相互作用保护了PZA的羰基,促进PZA分子通过N—H…N'在链中的组装,成功促使了PZA的γ晶型的成核。
虽然基于共晶设计的表面选择策略能够有效地对目标晶型进行调控,但是其表面的设计是建立在共晶数据的基础上,且形成的共晶须能够改变目标晶体的构型或者构象,这就限制了其在表面诱导成核多晶型中的应用。
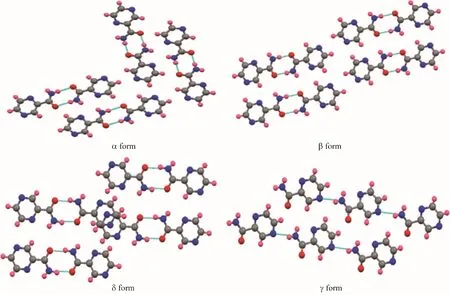
图10 吡嗪酰胺多晶型:4种堆积多晶型[71]Fig.10 Pyrazinamide polymorphic forms:four packing polymorphisms[71]
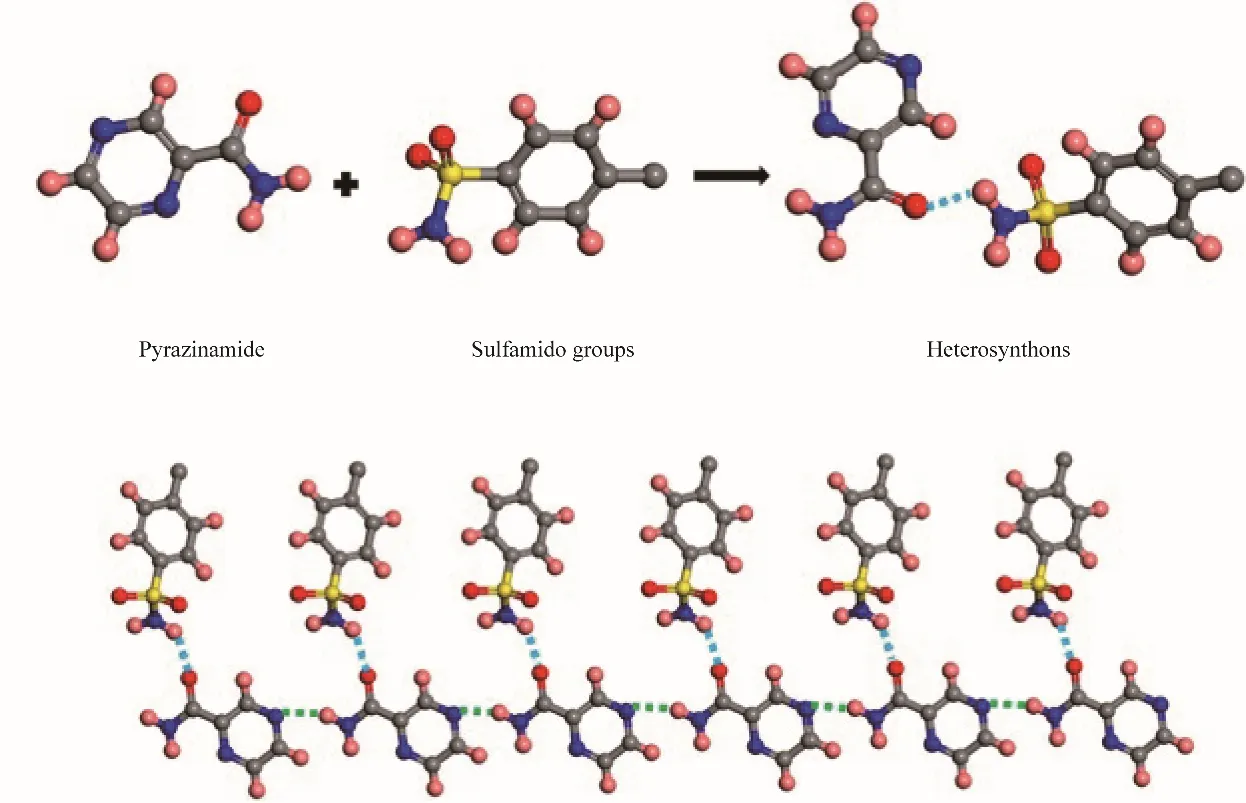
图11 酰胺基团的羰基与磺胺基团形成异相合成子[71]Fig.11 Heterosynthon diagram of the carbonyl moiety of the amide group and the sulfonamide moiety[71]
3.3 基于外延作用的表面设计
溶质分子在特定晶面的外延生长是有机小分子晶体调控晶型的重要作用机制,该策略的方法是使用生长较好的小分子晶体作为基底,晶体基底暴露的不同晶面可作为API 成核的位点,其在特定晶面的成核与生长可能是晶型调控的原因。由于溶剂体系不易选择,升华结晶往往是利用外延生长策略常采用的结晶方式,但适合升华结晶的研究体系相对匮乏,所以限制了此类研究的开展。
由于外延匹配强调晶体与基底的晶格匹配作用,采用结构相似的物质作为基底更容易发生外延作用。Srirambhatla 等[72]提出了一种基于同晶型模板基底诱导亚稳态晶型的方法,利用DHC−Ⅱ(双氢卡马西平晶型Ⅱ)薄膜多晶层作为模板表面,成功诱导产生CBZ−V(卡马西平晶型Ⅴ),而在非模板表面产生了CBZ−Ⅰ(卡马西平晶型Ⅰ)。随后他们将多晶DHC−Ⅱ模板诱导法应用于环庚米特(CYH),成功获得了一种新的晶型——CYH−Ⅲ(环庚米特晶型Ⅲ)。对三种物质的单晶结构进行解析发现,它们的单晶结构高度相似,这为利用外延作用调控亚稳晶型提供一种可行的办法。此外同一种模板晶体的不同晶型,对于结晶过程同样会具有不同的影响。Lee等[73]报道了结构相关的化合物甲灭酸(MFA)在氟灭酸(FFA)两种不同晶型(FFA−Ⅰ, FFA−Ⅲ) 单晶(100)面的外延生长行为,虽然外延晶体MFA 都表现出最稳定的晶型,但是在两种不同晶型的FFA 表面却表现出不同的结晶行为。在FFA−Ⅲ表面,MFA 的生长方向与表面彼此平行,而在FFA−Ⅰ表面,MFA 表现出两种不同的生长方向;MFA 晶体在FFA−Ⅰ表面表现出更快成核速度。晶格与构象匹配是MFA 在FFA 特定晶面成核以及成核速率不同的原因,这为通过外延作用调控晶型提供了一种新的思路。
基于结构相似性可以有效选择表面基底,但是外延作用由于存在不可预见性,导致有效的表面基底结构较少。Ward 等[32]开发了一种用于外延几何建模的软件——GRACE(晶体外延的几何实空间分析),通过比较基底的二维晶格参数和生长核的覆盖层的晶格参数,可以表征外延。这样的比较完全依赖于几何原理,提供了一种事先预测外延作用的可能性,并可对已发生的外延作用进行“外延评分”。这种探测外延关系的几何技术已经证明可以与势能计算所得到的结果相媲美,并提供了一种简便的途径来执行更多的计算,以获得等效的计算投资。利用GRACE 软件辅助计算,无论是以无机晶体还是有机小分子晶体作为基底方面,都得到了应用的范例[31−32,73]。
3.4 基于角向诱导的表面设计
多年以来,研究者们一直在探索通过控制异质表面的形貌来调控晶型,表面的划痕、沟槽、楔形能够显著提高成核速率,异质表面的孔道也被报道用于对晶型的调控,尤其是孔径小于100 nm 时,然而孔道形状的作用一直被忽视。对于孔道形状的影响缺乏系统的研究,很大程度上是因为制作具有可调几何形状的纳米孔的宏观材料面临挑战,尤其是孔径小于100 nm 的纳米孔。这主要是由于对于小分子晶体而言,分子聚集与定向排列往往是发生在几个纳米的尺寸上。为此,需要一种先进的制造技术来控制表面孔的几何形状和孔的大小,直至与成核相关的长度尺度,特别是使表面孔的尺寸从几纳米到数百纳米。纳米孔的密度需足够高,孔道数量充足,保证观察到的对成核的影响具有统计学意义[74]。为了满足这些要求,Trout 课题组[38,75]发明了一种“ 纳米压印光刻”(nanoparticle imprint lithography, NpIL) 技术,该技术利用纳米粒子作为模板用于制造具有各种形状的纳米孔阵列的聚合物表面,纳米孔大小从10纳米到数百纳米不等。
“角向诱导”的核心设计理念是计算API分子主要晶面之间的角度,并以此作为孔道形状的设计依据,利用NpIL技术在聚合物表面制备相应形状的孔道(图12),通过孔道的“约束效应”对API 的成核进行调控,10 nm 以下的孔隙往往不被考虑,主要是因为体积限制对成核的影响可能会掩盖孔隙形状的影响。Trout 课题组[38]将这种设计思路用于对甲灭酸的成核过程的研究,实验发现,在以羟甲基纤维素作为聚合物表面的情况下,甲灭酸的亚稳晶型Ⅱ仅出现在具有方形纳米孔的表面上,且这种表面促进成核的效果最强。
Trout 课题组[75]随后在对阿司匹林与吲哚美辛的研究中发现,在固定过饱和度下,聚乙烯醇薄膜由于具有良好的化学相互作用,显著缩短了成核诱导时间,当表面纳米压印的角度与不同晶面之间的角度匹配时,可进一步缩短成核诱导时间。同时γ晶型的吲哚美辛优先在65°与80°孔道的表面形成,且成核诱导期得到最大的缩短。

图12 聚合物表面的AFM图像[75]Fig.12 AFM images of polymer surface[75]
Diao 等[74]在对“角向成核”的研究中发现,聚合物薄膜表面与溶质分子的化学作用是诱导成核的先决条件,基于此,孔隙的几何约束作用会促进溶质分子的有序排列。他们以丙烯酸与二乙烯基苯交联的可与阿司匹林形成氢键的聚合物薄膜作为表面,发现圆形孔道阻碍了阿司匹林的成核,而六边形与方形的孔道却能有效降低阿司匹林的成核诱导期。他们认为有利的表面−溶质相互作用提高了表面附近的溶质浓度,表面和溶质之间的分子识别使得富集的溶质分子定向顺序,这两种作用都能促进阿司匹林的成核;其次孔隙的几何约束进一步增强了溶质分子在表面的取向顺序,有利于溶质分子在成核过程重新排列。当几何角度作用下的分子取向与晶体相似时,成核速率得到提高。根据他们的假设,有利的表面−溶质的相互作用是促进成核的先决条件。随后他们选择了对于阿司匹林成核无影响的聚合物薄膜作为异质表面,发现在其表面上制备的六方形与方形的孔道并不能促进阿司匹林的成核,验证了表面−溶质的相互作用对于成核的重要作用。
4 结论与展望
利用表面诱导成核的方法成功发现了许多药物的新晶型,同时在亚稳晶型的调控方面也卓有成效,丰富了晶型的调控方法。异质表面通过化学作用与几何作用来调控多晶型的成核,相比于几何作用,化学作用具有明显的优势。现阶段异质表面的选择主要聚焦在聚合物、自组装单层膜、有机小分子晶体、凝胶上。在聚合物方面,因其可通过选择不同的单体合成具有不同侧链官能团的功能化表面或是借助于纳米压印光刻等先进技术赋予其不同的表面形貌而得到广泛研究;SAMs 由于其具有高度有序致密的功能化单分子层,基于各种分子间作用力的化学作用也得到一定研究;而在有机小分子晶体方面,外延生长的研究多是集中在这类异质表面上,但溶剂体系的制约限制了它在诱导药物多晶型的应用;凝胶相结晶作为一种新兴的结晶方法目前还存在一定的盲区,但是随着研究的深入,将会在药物多晶型成核方面得到更多的应用。虽然利用表面诱导药物多晶型成核已经走过二十多年的历史,目前还存在诸多的问题,如:
(1)表面诱导多晶型成核的机理还不明确,分子模拟可为机理的明确提供新的见解,但模拟大多忽视了溶剂对于表面诱导成核的影响;
(2)异质表面的选择策略还不明确,表面的选择更多是一个“试错”的过程,需要耗费大量的人力、物力,如何高效设计与选择合适的异质表面成为亟待解决的问题;
(3)目前利用表面诱导策略调控药物多晶型成核还停留在实验室阶段,未有工业化应用的实例,这主要是由于表面诱导多晶型成核的机理还不明确以及大尺度下流体力学的改变会对成核造成很大的影响。
随着检测与分析技术的进步,表面诱导成核的机理会逐渐完善;计算机技术的发展将有助于利用分子模拟确定溶质分子与异质表面之间的相互作用关系,用于指导表面的设计,使表面的设计与选择更加高效。未来,通过表面诱导成核将实现药物新晶型的筛选、多晶型的精准调控和制备。
