6-无氟喹诺酮类抗菌药物药理学及临床研究进展
2021-02-22陈虹彤卢芸李国庆游雪甫杨信怡
陈虹彤,卢芸,李国庆,游雪甫,杨信怡
·综述·
6-无氟喹诺酮类抗菌药物药理学及临床研究进展
陈虹彤,卢芸,李国庆,游雪甫,杨信怡
100050 北京,中国医学科学院北京协和医学院医药生物技术研究所抗感染药物研究北京市重点实验室
早期喹诺酮品种如萘啶酸、吡哌酸化学结构中均不含氟,但自 1984 年日本杏林制药株式会社开发的首个具有6-氟-7-哌嗪基结构的喹诺酮药物——诺氟沙星(norfloxacin,氟哌酸)成功上市以来,以 4-喹酮为母核(结构见图 1),6 位碳(C-6)有氟原子(F)取代为特征的 6-氟喹诺酮类抗菌药因抗菌谱广、疗效可靠、使用方便等优点,一直是新药开发的主流[1-2]。以我国为例,截至 2017 年,上市的17 个全身用喹诺酮药物品种均为 6-氟喹诺酮[3];而近30 年来,美国 FDA 批准的喹诺酮新药品种中 6-氟喹诺酮药物亦占绝对多数[4]。
然而,随着氟喹诺酮药物在临床领域的广泛使用,除因选择压力导致细菌耐药性问题日益严重,不断报道的用药安全问题也逐渐凸显。少数患者接受某些氟喹诺酮品种治疗后,会出现罕见而严重的不良反应,如肌腱炎/肌腱断裂、中枢/周围神经病变、QT 间期延长、光毒性、血糖异常、肝炎、溶血性贫血等。由于这些不良反应对人体具有致残或不可逆性风险,氟喹诺酮的用药安全性问题多次在公众中引起广泛关注和讨论,并导致部分品种终止上市或退市[5]。例如,1999 年,曲伐沙星(trovafloxacin)上市仅一年即因严重肝毒性被美国限制使用,随后被欧盟终止上市;2000 年前后,司帕沙星(sparfloxacin)由于光毒性问题,在欧美的使用严格受限;2007 年,上市多年的加替沙星(gatifloxacin)因诱发严重糖代谢紊乱而从美国退市,如此等等[6-7]。2016年7月,美国 FDA 建议对静脉和口服氟喹诺酮类药物的说明书进行修改,警示这类药物全身性应用可能引发致残和永久性损害的潜在风险,并限制其在非严重感染患者中的使用[4]。鉴于此,寻找和开发更为安全、有效的新型喹诺酮品种一直是医药工作者不懈努力的目标。

图 1 喹诺酮母核 4-喹酮结构图
1992 年,Ledoussal 等[8]首先报道了喹诺酮母核 C-6位无氟(或脱氟),但具有良好体外抗菌、抑酶(细菌 DNA促旋酶和/或拓扑异构酶 IV)活性的 6-无氟喹诺酮系列化合物,并指出 C-6 位氟代并非喹诺酮类高抗菌活性的前提,且 C-6 位脱氟后致遗传毒性相对减弱,较氟喹诺酮类安全性更高。因而提出只要母核 7 位碳(C-7)上连有适宜取代基团,完全可能筛选出抗菌活性可与 6-氟喹诺酮媲美的新型 6-无氟喹诺酮。此后,6-无氟喹诺酮类的开发价值逐渐受到各国药物研发机构的重视,一系列 8-卤素-6-H 喹诺酮类化合物获得专利,继而 8-甲基-6-H 喹诺酮类及 C-7 位上有独特芳基取代基的新型 6-无氟喹诺酮类药物先导物相继被报道[1]。2007 年,富山化学工业/大正制药联合开发的首个 6-无氟喹诺酮类抗菌药——加雷沙星在日本上市。该药在临床应用中表现出优良的抗菌疗效及较低的不良反应发生率。此后,又有多个在研 6-无氟喹诺酮类药物品种相继进入临床或临床前研究阶段,这些品种的基本情况参见表 1。鉴于 6-无氟喹诺酮类药物在临床上具有广阔的应用发展空间,本文针对新型 6-无氟喹诺酮类抗菌药物的最新研究进展作一综述,为医药工作者提供参考。

表 1 上市及在研 6-无氟喹诺酮类药物[9]
1 6-无氟喹诺酮类药物作用及耐药机制
6-无氟喹诺酮类药物的细菌作用靶标与氟喹诺酮类无别,亦作用于拓扑异构酶 II(DNA 促旋酶)和拓扑异构酶 IV。研究显示,6-无氟喹诺酮类药物对细菌靶酶的选择作用因细菌种类而异[10]。例如,针对大肠埃希菌,6-无氟喹诺酮类药物主要抑制 DNA 促旋酶,且对一些存在位点突变的 DNA 促旋酶也具有抑制活性,但对拓扑异构酶 IV 无明显抑制作用。针对金黄色葡萄球菌(下文均简称金葡菌),6-无氟喹诺酮类药物则表现出双酶抑制作用,故金葡菌中单一靶酶编码基因发生突变并不必然导致耐药。在肺炎链球菌、结核分枝杆菌等病原菌中,6-无氟喹诺酮作用的主要靶标仍为 DNA 促旋酶[1, 11]。
目前认为,细菌对 6-无氟喹诺酮主要通过两种机制产生耐药:①编码靶蛋白 A、B 亚基的细菌染色体基因(、、、)发生突变;②减少摄取(膜孔蛋白突变)或增加外排(外排泵蛋白过度表达)以减少药物在细胞内的积累。革兰氏阳性菌对喹诺酮耐药性主要与、编码基因的突变及部分外排泵蛋白的过表达相关[12]。
2 已上市6-无氟喹诺酮类药物
2.1 加雷沙星
加雷沙星(garenoxacin,BMS-284756,T-3811,佳诺沙星,加诺沙星)英文商品名 Geninax。其药用形式为甲磺酸盐水合物,相对分子量(MW)426.42,由日本富山化学工业株式会社与大正制药株式会社联合成立的大正富山制药株式会社开发,富山化学工业株式会社生产,2007年8月经日本劳动厚生省批准上市。后授权美国先灵葆雅公司(Schering-Plough Co.,Ltd,现已并入美国默沙东公司)负责在中、日、韩以外地区的上市和推广。目前,其在欧美的上市申请尚未获批准。加雷沙星化学结构式见图 2,其 C-7 位侧链连接一个含芳环结构的 5-异吲哚啉基而非常见的环胺基,目的在于保持对革兰氏阴性菌良好活性的基础上,增强对革兰氏阳性菌,尤其是对肺炎链球菌的抗菌活性。该药既可口服又可注射给药[13]。
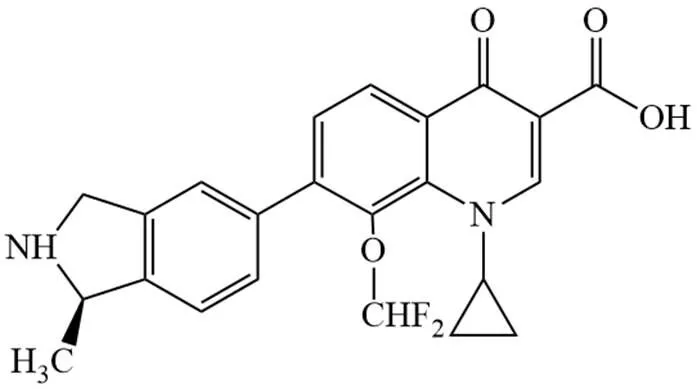
图 2 加雷沙星化学结构
加雷沙星对葡萄球菌(包括甲氧西林耐药菌)、链球菌属(包括环丙沙星不敏感、青霉素耐药的肺炎链球菌)、粪肠球菌等革兰氏阳性菌都有良好体外抗菌活性[14]。对环丙沙星敏感、不敏感金葡菌 MIC90分别为 0.03 和4 mg/L[15]。对甲氧西林敏感和耐药金葡菌(MSSA/MRSA)MIC90分别为 0.03 和 2 mg/L。且加雷沙星对部分葡萄球菌有快速杀灭作用,10 × MIC 浓度下,2 ~ 4 h 可杀减活菌数 3 log10[16]。一项药敏研究报道,对受试 35 株青霉素敏感肺炎链球菌(PSSP),几种喹诺酮抗菌药的体外抗菌活性排序:加雷沙星(MIC90= 0.12 mg/L)> 莫西沙星(MIC90= 0.25 mg/L)> 左氧氟沙星(MIC90= 1 mg/L)> 环丙沙星(MIC90=2 mg/L)。0.25 mg/L 加雷沙星可以抑制 90% 的酿脓链球菌,其抗菌活性不受细菌对大环内酯类抗生素敏感性的影响[17]。但加雷沙星对链球菌及肠球菌杀菌速度较慢,在10 × MIC 浓度下,> 6 h 才可减少活菌数 3 log10[16]。
对革兰氏阴性菌,加雷沙星对肠杆菌科细菌和大多数非发酵菌有活性(MIC ≤ 4 mg/L),但总体活性弱于环丙沙星[14]。对巴斯德菌(包括产 β-内酰胺酶菌株)的 MIC ≤0.06 mg/L,优于左氧氟沙星和莫西沙星[18]。在临床剂量水平可有效抑制空肠弯曲杆菌(MIC50= 0.03 mg/L)、幽门螺杆菌(MIC90= 0.008 mg/L)、军团病杆菌属[19]。一项研究显示,在 10 × MIC 浓度下,受试的加雷沙星和氟喹诺酮类使活菌数降低 3 log10所需时间 < 2 h,而 β 内酰胺类抗生素则≥ 6 h[16]。
在现有喹诺酮类药物中,加雷沙星对厌氧菌抗菌谱最广,绝大部分厌氧菌对其敏感,同时也是对难培养病原体(支原体、脲原体、衣原体、环丙沙星非敏感性淋球菌)活性最强的喹诺酮品种[14]。其对肺炎支原体 FH 的 MIC 为 0.0313 mg/L,是莫西沙星、加替沙星、左氧氟沙星 MIC 的 1/2、1/4、1/16[20]。
用幼年比格犬模型评价加雷沙星的关节毒性,结果发现,每天 1 次,连续 7 天静脉给药 30 mg/kg,加雷沙星组幼犬组织病理学检测未见关节毒性,而环丙沙星、诺氟沙星给药组幼犬均检测到多部位关节软骨病变。该研究中加雷沙星0→∞为 164 μg·h/ml,高于环丙沙星和诺氟沙星。每天 1 次,连续 7 天口服给药 50 mg/kg,加雷沙星、环丙沙星、诺氟沙星在雄性幼犬血浆中浓度分别为 14.4、3.72、1.71 mg/L,在雌性幼犬中分别为 13.7、5.67、1.90 mg/L;加雷沙星在雄性和雌性幼犬膝关节滑液、软骨、滑膜标本中的浓度分别为环丙沙星和诺氟沙星浓度的 2.0 ~ 6.5 倍和 1.5 ~ 3.3 倍。组织病理学改变程度:环丙沙星> 诺氟沙星> 加雷沙星。这反映加雷沙星的血浆及关节中浓度虽明显高于环丙沙星和诺氟沙星,但关节毒性却远低于二者[21]。
加雷沙星具有浓度依赖性药代动力学特征,血清蛋白结合率为 78%,主要经肾脏及粪便排泄[22]。该药口服吸收快,达峰时间为 1.13 ~ 2.5 h,生物利用度为 43% ~ 96%,半衰期为 13.3 ~ 17.8 h,30% ~ 50% 药物以原形从尿中排出[23]。对健康受试者,每日 200 mg/次给药,max和0-24分别为 4.89 mg/L 和 48.9 μg·h/ml。对于肾功能正常及中度肾功能不全的呼吸道细菌感染患者,加雷沙星推荐剂量为400 mg/d[24],但对于体重 < 40 kg 或肌酐清除率 < 30 ml/min的患者,加雷沙星推荐剂量为 200 mg/d[25]。在经维持性血液透析(MH)治疗的肾功能不全患者中,口服加雷沙星 200 mg/d,max降低 40%,为(3.0 ± 1.12)mg/L,0-24参数与健康受试者相近,为(40.7 ± 16.7)μg·h/ml[25]。
加雷沙星甲磺酸水合物为其口服剂型。作为广谱抗菌药,该药用于社区获得性呼吸道感染、急性上颚窦炎、泌尿生殖系统、皮肤和软组织感染等疾病的临床治疗,其对很多耐药菌感染也具良好的抗菌疗效[26]。在II期、III期临床试验中,加雷沙星对革兰氏阳性菌和革兰氏阴性菌的总清除率分别为 97.4% 和 92.7%[26]。在受募参加加雷沙星临床应用安全性评价的 702 位受试者中,总不良反应发生率为 34.8%(244/702),药物相关不良反应发生率为 12.7%(89/702),均为轻、中度不良反应,其中腹泻发生率最高,其次为恶心及头痛[26-27]。Kohno 等[28]的研究表明,对早期采用静脉注射舒巴坦/氨苄西林治疗的社区获得性肺炎(CAP)患者,转用口服加雷沙星 400 mg/d,疗效不劣于前者的持续治疗。且转用加雷沙星后可缩短患者住院时间,减少医疗支出。临床试验 UMIN000012627 中评价了口服加雷沙星(400 mg/d)与西他沙星(100 mg/d)治疗≥ 65 岁肺炎患者的有效性与安全性。结果显示,两药的临床治愈率分别为 88.9% 和 88.5%,药物相关不良反应发生率分别为 27.9% 和 20.7%,无显著性差异,两者最常见不良反应均为肝功能异常。该项研究最终评定认为两药均有较高有效性及安全性,且未对老龄患者提出剂量调整的需求[29]。
据 2009年10月– 2011年7月加雷沙星在日本上市后的监管统计,口服该药对非典型肺炎疑似患者和确诊患者有效率分别为 94.8%(55/58)和 92.3%(12/13),药物相关不良反应发生率为 4.8%(5/105),消化道疾病、感染、神经系统疾病、皮肤和皮下组织疾病发生率分别为 2.9%、1.0%、1.0%、1.0%[30];在印度开展的IV期临床试验中,461 名社区获得性呼吸道感染患者连续 5 ~ 14 天口服甲磺酸加雷沙星片剂 2 × 200 mg/d。53 名(11.5%)患者在治疗期间出现不良反应,主要为神经系统疾病和消化系统疾病,其中最常见的不良反应包括腹部不适(3,0.6%)、头晕(10,2.1%)、头痛(10,2.1%)、食欲下降(5,1.1%),未出现严重不良反应[31]。
2.2 奥泽沙星
奥泽沙星(ozenoxacin,T-3912)由 Toyama 化学公司研发,并授权日本 Maruho 公司和西班牙制药公司格鲁波•费雷尔国际公司(Grupo Ferrer Internacional S.A.)进行全球推广。2017年12月11 日,美国 FDA 批准 Xepi™(ozenoxacin 1%)上市,作为局部应用的乳膏,用于成人和 2 个月及以上儿童患者治疗金葡菌或化脓链球菌引起的脓疱疮(又名脓疱病、黄水疮)[32]。本品结构如图 3 所示,其 C-7 的吡啶环取代可增强抗革兰氏阳性菌的活性。

图 3 奥泽沙星结构
奥泽沙星可同时抑制细菌 DNA 促旋酶 A 和拓扑异构酶 IV,且不易受某些外排泵活性蛋白的影响,因而被认为较能有效防止细菌耐药性发展[33-34]。奥泽沙星抗菌活性较强,可能与其能快速入胞渗透并维持较高胞内浓度有关,也一定程度反映出奥泽沙星对某些外排泵蛋白不敏感[33]。对于在喹诺酮抗性决定区中存在突变的菌株,奥泽沙星诱导自发抗性突变的频率低[35]。另有研究显示奥泽沙星体外抗菌活性不受阳离子浓度、细菌接种量、培养时间、CO2等因素的影响,且在酸性 pH 环境(皮肤及软组织环境为酸性)下仍具良好活性,而环丙沙星、诺氟沙星等氟喹诺酮药物在酸性环境下失活[36],这些体外证据均支持该药开发成为抗皮肤感染治疗用药。
目前,奥泽沙星体外抗菌活性的研究主要针对皮肤及软组织感染病原菌进行。奥泽沙星对 MSSA、耐氧氟沙星 MRSA、表皮葡萄球菌(下文称表葡菌)、耐氧氟沙星表葡菌、耐青霉素肺炎链球菌(PRSP)、痤疮丙酸杆菌的 MIC90分别为 0.00625、0.2、0.0125、0.2、0.05、0.05 mg/L,体外抗菌活性是那氟沙星(nadifloxacin)、氧氟沙星、左氧氟沙星、氯林可霉素、红霉素、庆大霉素的 4 ~ 1600 倍;对 PSSP 的 MIC90= 0.1 mg/L,活性弱于氯林可霉素(MIC90= 0.025 mg/L),但是上述其他药物的 4 ~ 128 倍;对酿脓链球菌的活性与红霉素相近,MIC90= 0.05 mg/L,活性是上述其他药物的 2 ~ 256 倍;对铜绿假单胞菌 MIC90=6.25 mg/L,活性弱于氧氟沙星(MIC90= 3.13 mg/L)和左氧氟沙星(MIC90= 3.13 mg/L)[37];奥泽沙星对左氧氟沙星敏感/耐药的金葡菌和链球菌都具有良好的抗菌活性,MIC90≤ 0.5 mg/L[38]。此外,奥泽沙星对 grlA 突变金葡菌也具有活性。该药与其他类别抗革兰氏阳性菌药物无交叉耐药性。金葡菌 SA113 和痤疮丙酸杆菌 JCM 6425 针对奥泽沙星的自发突变率不高,分别 < 1.7 × 10-9和 < 2.0 × 10-9[37, 39]。对左氧氟沙星敏感及耐药痤疮丙酸杆菌,奥泽沙星的最低杀菌浓度(MBC)分别为 0.06 ~ 8 mg/L 和 0.5 ~ 4 mg/L,且对左氧氟沙星敏感痤疮丙酸杆菌具有持久的抗生素后效应(PAE,3.3 ~ 17.1 h),但对左氧氟沙星耐药菌不具有 PAE[40]。总之,这些体外药效数据均支持奥泽沙星用于寻常痤疮的治疗。
Ames 试验(污染物致突变性检测)、小鼠淋巴瘤细胞试验、大鼠微核试验中,奥泽沙星均未表现出遗传毒性[32]。幼鼠及幼犬口服奥泽沙星耐受性好,未出现软骨毒性或靶器官相关毒性[41]。在金葡菌引起的皮肤感染小鼠模型中,局部应用奥泽沙星减少细菌载量的最有效浓度为 1% 和 2%。该模型的实验治疗中,1% 奥泽沙星软膏和乳膏治疗组细菌清除率分别为 47% 和 53%,明显高于 2% 莫匹罗星软膏和 1% 瑞他莫林(又名瑞他帕林)软膏治疗组的清除率(分别为 28% 和 31%),各给药组均未见局部不良反应[42]。
I 期临床研究中,奥泽沙星 1% 及 2% 软膏体表局部给药,无透皮吸收,在表皮层中含量低但在角质层中含量高。无软骨毒性,未见光毒性、光过敏、接触性过敏等反应[33, 43]。在 4 个 I 期临床研究中,健康受试者局部用药对奥泽沙星无系统吸收,且耐受性良好,最常见的药物相关不良事件是用药部位反应(红斑和瘙痒)[44]。因奥泽沙星临床用药方式为局部用药,基本无系统吸收,故后续临床研究中均未开展药物分布、消除、排泄相关研究[32, 45]。
在III期临床脓疱疮患者治疗试验中,连续 5 d、2 次/d 在患者感染部位给予奥泽沙星1% 乳膏、安慰剂乳膏或瑞他莫林 1% 软膏治疗。在 3 ~ 4 d 后奥泽沙星组微生物清除率高于瑞他莫林组(74.7% vs. 60%),这表明与后者相比,奥泽沙星能更快速地清除细菌[46]。另一治疗脓疱疮的III期临床试验(NCT02090764)显示,与安慰剂(溶媒)乳膏相比,采用奥泽沙星 1% 乳膏剂 5 d、2 次/d 局部治疗可显著提高临床成功率(54.4% vs. 37.9%)及微生物清除率(87.2% vs. 63.9%)。药物安全性较高,患者耐受良好,总不良反应发生率 3.9%(8/206),药物相关不良反应发生率 0.5%(1/206),未见严重不良反应[47]。尽管奥泽沙星体表杀菌活性较强,尤其是对革兰氏阳性菌活性突出,且临床试验显示其安全有效,但由于其成本较高,且缺少与临床推荐用药的直接比较性试验数据,目前尚不太可能替代莫匹罗星成为治疗脓疱疮的一线药物,但可作为莫匹罗星或瑞他莫林治疗无效的耐药性脓疱疮的备选药[48]。
2.3 奈诺沙星
奈诺沙星(nemonoxacin,Taigexyn,TG-873870)是宝洁公司开发的新型 C-6 位无氟喹诺酮药物,口服可用于 CAP、糖尿病足溃疡感染、皮肤和软组织感染的治疗,静脉主要用于 CAP 治疗。中国台湾于 2014年3月13 日已批准将口服苹果酸奈诺沙星胶囊(Taigexyn)用于治疗成人 CAP[49],2016年5月27 日,该药在中国大陆获得 CFDA 的批准文号及新药证书。奈诺沙星结构见图 4,该药同样选择性抑制细菌 DNA 促旋酶 A 和拓扑异构酶 IV。

图 4 奈诺沙星结构
奈诺沙星抗菌谱广,多项研究显示,奈诺沙星对甲氧西林敏感表葡菌(MSSE,MIC90= 0.5 mg/L)、甲氧西林耐药表葡菌(MRSE,MIC90= 2 mg/L)、MSSA(MIC90≤0.03 mg/L)、MRSA(含环丙沙星敏感 MRSA,MIC90≤0.03 mg/L)、左氧氟沙星敏感金葡菌(MIC90= 0.06 mg/L)、社区获得性 MRSA(CA-MRSA)(MIC90= 0.06 ~ 0.5 mg/L)、肺炎链球菌(含 PRSP,MIC90= 0.015 mg/L)等革兰氏阳性菌均具较好抗菌活性[50-51],但对环丙沙星耐药 MRSA(MIC90= 1 mg/L)、万古霉素中介 MRSA(MIC90= 2 mg/L)、达托霉素非敏感 MRSA(MIC90= 1 mg/L)、院内获得性 MRSA(HA-MRSA,MIC90= 16 mg/L)、万古霉素敏感肠球菌(粪肠球菌 MIC90= 2 mg/L,屎肠球菌MIC90= 4 mg/L)、万古霉素耐药肠球菌(粪肠球菌 MIC90= 4 mg/L;屎肠球菌 MIC90= 16 mg/L)抗菌活性较弱[49]。另外,奈诺沙星对诺卡菌属也有良好的抗菌活性,对巴西诺卡菌、星形诺卡菌、其他诺卡菌种属的 MIC90分别为 0.5、8、2 mg/L,活性高于吉米沙星(MIC90:1、16、16 mg/L)和左氧氟沙星(MIC90:8、32、16 mg/L)[52]。对于革兰氏阴性菌,奈诺沙星对流感嗜血杆菌的活性优于环丙沙星、左氧氟沙星、莫西沙星,4 个药的 MIC90分别为 4、16、8、> 8 mg/L;但对鲍曼不动杆菌、铜绿假单胞菌、大部分肠杆菌属细菌活性较差,MIC90均 > 16 mg/L[53]。奈诺沙星治疗结核病(TB)的作用有限,对耐多药结核分枝杆菌(MDR-Mtb)和非耐多药结核分枝杆菌的 MIC90分别为 > 32 mg/L 和 16 mg/L[54]。另外,奈诺沙星对非典型病原体,如沙眼衣原体、肺炎衣原体也具有良好活性(MIC90= 0.06 mg/L)[55]。
在小鼠全身感染模型中,奈诺沙星对革兰氏阳性球菌 MSSA、MRSA、左氧氟沙星耐药的甲氧西林耐药头状葡萄球菌(levofloxacin-resistant MRSC)、青霉素中介肺炎链球菌(PISP)、PRSP、粪肠球菌感染小鼠的 ED50分别为 2.08、2.59、2.52、5.47、3.68 ~ 5.28、8.48 ~ 15.16 mg/kg,疗效明显高于左氧氟沙星(ED50分别为 5.02、8.45、4.32、19.14、19.82 ~ 22.01、17.47 ~ 26.89 mg/kg)。但对大肠埃希菌所致感染,本品相对左氧氟沙星体内抗菌疗效较弱(ED50,3.13 ~ 5.28 mg/kg vs. 0.68 ~ 0.97 mg/kg)。在鼠肺感染模型中,奈诺沙星对 PRSP 和肺炎克雷伯菌所致感染均有抗菌效果。其中,对肺组织中 PRSP 的清除作用高于左氧氟沙星(20 mg/kg:降至 2.93 log CFU/g vs. 降至 4.34 log CFU/g;10 mg/kg:降至 4.01 log CFU/g vs. 降至 4.76 log CFU/g),但对肺炎克雷伯菌的清除作用低于左氧氟沙星(20 mg/kg:降至 4.49 log CFU/g vs. 降至 3.02 log CFU/g;10 mg/kg:降至 5.88 log CFU/g vs. 降至 3.33 log CFU/g)。在鼠上行尿路感染模型中奈诺沙星对肾组织中大肠埃希菌的清除作用弱于左氧氟沙星(4 mg/kg:降至 4.60 log CFU/g vs. 降至 3.47 log CFU/g;2 mg/kg:降至 4.85 log CFU/g vs. 降至4.00 log CFU/g)[56]。
健康受试者口服奈诺沙星的I期临床试验显示,奈诺沙星口服吸收快速,血浆浓度在1 ~ 2 h 内达峰,表观分布容积为 2.78 ~ 5.35 L/kg,生物利用度大于 95%,与左氧氟沙星、莫西沙星相近。血浆蛋白结合率约 16%,低于市场上现有喹诺酮类药物。摄食会显著降低其max和,并使达峰时间由 1.14 h 延长至 3.64 h。消除半衰期为 10 ~ 12 h,奈诺沙星主要经肾脏清除,随尿液排出,35% ~ 65% 的药物以原型药排出。奈诺沙星不是人体肝 CYP3A4 的抑制剂或诱导剂[57-58]。静脉注射奈诺沙星的I期临床试验 NCT01944774 显示,健康人体对静脉注射该药的最大耐受剂量为 1250 mg,可接受注射速度范围为 0.42 ~ 5.56 mg/min。0-72 h和0-∞在 250 ~ 750 mg 给药剂量范围内呈线性剂量反应关系[59]。
II期(NCT01537250)临床试验共纳入 192 位中国 CAP 成年患者,对奈诺沙星口服500 mg/d 及 750 mg/d 的安全性及临床有效性进行了评价。试验期间,肺炎链球菌、流感嗜血杆菌、肺炎支原体、肺炎衣原体、嗜肺军团菌感染患者均获成功治愈。750 mg 及500 mg 奈诺沙星治疗组中药物相关不良反应发生率分别为 35.6% 和 30.6%,多数为轻度、短暂不良反应,主要为胃肠道反应(如恶心、呕吐)、暂时性中性粒细胞减少、肝转氨酶升高等,未见与药物相关的严重不良事件[60]。III期(NCT01529476)非劣效性临床试验进一步评价了 CAP 患者口服 500 mg 奈诺沙星的疗效及安全性,共包含 527 名患者(奈诺沙星治疗组 356 人,阳性对照药左氧氟沙星治疗组 171 人)。总体而言,奈诺沙星非劣效于左氧氟沙星(500 mg),两者临床治愈率分别为94.3% 和 93.5%,微生物学清除率分别为 92.1% 和 91.7%,不良反应发生率分别为 33.1% 和 33.3%,其中药物相关不良反应发生率分别为 19.4% 和 17.5%,大部分为轻至中度不良反应,奈诺沙星与左氧氟沙星治疗组药物相关不良反应无显著差异,主要包括丙氨酸转氨酶升高(5.1% vs. 4.1%)、白细胞减少(2% vs. 1.2%)、恶心(3.1% vs. 1.8%)、呕吐(1.7% vs. 2.3%)。这些结果与II期 CAP 临床试验结果相近[61]。其他III期非劣效性试验(NCT02205112、NCT03551210)中,受试者中绝大部分为老年患者,这些试验也进一步证实了奈诺沙星在治疗成人 CAP 中的安全性和有效性[62-64]。
3 在研 6-无氟喹诺酮类药物
3.1 DX-619
DX-619是日本第一三共株式会社研发的 6-无氟喹诺酮品种,C-7 位有氨基环丙基吡咯烷基,C-1 位有氟化的环丙基,结构如图 5。
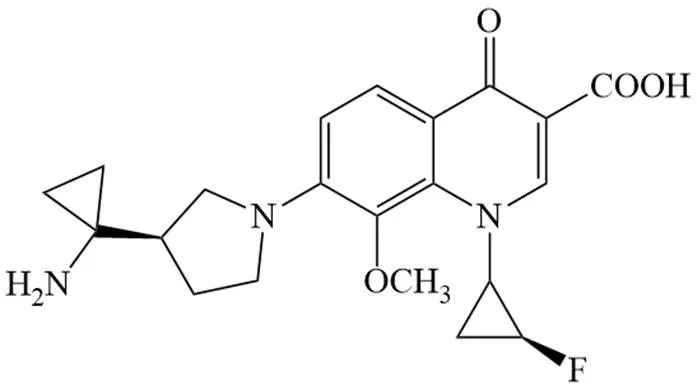
图 5 DX-619 结构
DX-619 对包括喹诺酮耐药菌株在内的革兰氏阳性和阴性菌具有广谱抗菌活性,尤其对耐多药革兰氏阳性球菌表现出良好抗菌活性[65]。对于革兰氏阳性菌,DX-619 对金葡菌和凝固酶阴性葡萄球菌的 MIC50/MIC90分别为 0.06/0.5 mg/L 和 0.016/0.125 mg/L。其中,对喹诺酮耐药葡萄球菌的 MIC50/MIC90为 0.125/0.5 mg/L,低于西他沙星(0.5/4 mg/L)、莫西沙星(2/8 mg/L)、加替沙星(4/16 mg/L)、左氧氟沙星(16/ > 32 mg/L)、环丙沙星(> 32/ > 32 mg/L)。其对 MRSA 也具有较好抗菌活性,MIC50/MIC90为 0.125/1.0 mg/L[65-66]。对青霉素敏感和非敏感性肺炎链球菌、粪肠球菌、屎肠球菌,其 MIC90分别为 0.016、0.062、0.25、0.5 mg/L;对突变的肺炎链球菌有很好的抗菌活性并且受突变影响较小,可能会成为该菌所致呼吸道感染的有效药物[67]。对于革兰氏阴性菌,DX-619 对肠杆菌科细菌的活性与莫西沙星和加替沙星相当,且对葡萄糖非发酵菌的活性强于后两药,对肠杆菌科细菌(大肠埃希菌除外)和非发酵葡萄糖杆菌的 MIC90≤ 4 mg/L。DX-619 对铜绿假单胞菌、鲍曼不动杆菌、噬麦芽寡养单胞菌的 MIC90在 0.5 ~2 mg/L[65]。对包括耐药菌株在内的幽门螺旋杆菌,DX-619 的 MIC50/MIC90为 0.008/0.06 mg/L,优于羟氨苄青霉素、左氧氟沙星。DX-619 的 MBC/MIC 为 1 ~ 2,杀菌作用与左氧氟沙星等氟喹诺酮类药物相当[68]。DX-619 对拟杆菌属、普氏菌属、梭杆菌属、微单胞菌属、放线菌属、梭菌属等厌氧菌均有较高抗菌活性,MIC50/MIC90范围在 0.03 ~ 0.25/ < 0.03 ~ 1 mg/L。DX-619 对亚胺培南耐药的杆菌属也具有很好抗菌效果,MIC50/MIC90为 0.25/1 mg/L[69]。
对于由 PSSP 和 PRSP 引起的肺炎感染小鼠,DX-619 可显著降低感染小鼠肺部细菌数。对 PSSP 引起的肺炎感染小鼠,DX-619、西他沙星、环丙沙星三个药物组(10 mg/kg,2 次/d)与生理盐水(2 次/d)对照组小鼠肺部细菌计数分别为 1.75、1.92、6.48、7.57 log10CFU/肺,对 PRSP 引起的肺炎感染小鼠,肺部细菌计数分别为 1.73、2.28、4.61、5.54 log10CFU/肺。DX-619 肺部/MIC 显著高于西他沙星、环丙沙星组,分别为 171.0、21.92、1.22[70]。DX-619 治疗 MRSA、万古霉素中介金葡菌(VISA)所致血源性小鼠支气管肺炎的研究中,DX-619 使 MRSA 感染小鼠的肺组织细菌计数明显减少。100 mg/(kg·d)、2 次/d、连续 7 d 注射 DX-619,与对照组肺部细菌计数分别为 2.91 及7.97 log10CFU/肺。同时,DX-619 可显著提高VISA 感染小鼠存活率(DX-619 vs. 对照组:90% vs. 45%),且组织病理学检查显示 DX-619 治疗组炎症较轻,反映该药具有治疗血源性支气管肺炎的潜力[71]。有报道显示,氧氟沙星对幼年大鼠具有软骨毒性,而 DX-619 对幼鼠无软骨毒性或毒性反应较低[72]。另有研究表明,治疗剂量内,DX-619 可引起血清肌酸酐水平升高,其机制与 DX-619 是肾阳离子转运体的抑制剂有关,其具有抑制 hOCT2、hMATE1、hMATE2-K 的作用,从而抑制肾小管分泌肌酸酐,导致血清肌酸酐水平升高[73-74]。
I 期临床研究中,连续 4 d 静脉注射 DX-619 800 mg、1 次/d,对健康受试者的肾小球滤过率无影响,且受试者血清肌酐水平的升高及肌苷酸清除率的降低均具可逆性,故现认为 DX-619 不会引起肾毒性[75]。但本品在完成了静脉注射 I 期临床试验后由于某些不明原因停止了开发,目前研究进展不明。
3.2 PGE 9262932、PGE 4175997、PGE 9509924
PGE 9262932、PGE 4175997、PGE 9509924 是由宝洁公司开发的系列 6-无氟喹诺酮类药物,结构如图 6 ~ 8 所示。体外条件下,三种药物抗菌活性排序为:PGE 9262932 > PGE 4175997 > PGE 9509924[76]。研究表明,这三种药抗菌谱广,对大多数革兰氏阳性菌活性高于曲伐沙星,但对肠杆菌科细菌活性较低,对假单胞菌属无明显活性[77]。关于 PGE 9262932 的研究显示,在受试 43 种 600 株临床分离菌(包括革兰氏阳性菌和阴性菌)中,85.3% 能被≤ 1 mg/L 的 PGE 9262932 抑制,其药代动学性质与已有的氟喹诺酮相似,但毒性低于相应氟喹诺酮[78]。
对革兰氏阳性菌,PGE 9262932、PGE 4175997、PGE 9509924对肺炎链球菌和其他链球菌的抗菌活性较强,是已知氟喹诺酮类药物的 2 ~ 128 倍,抗肠球菌活性与氟喹诺酮类药物相似[76]。对葡萄球菌属的活性与曲伐沙星相近,对曲伐沙星耐药葡萄球菌的 MIC 低于氟喹诺酮类药物。另外,三药对 MRSA 表现出快速杀灭作用,对大多数粪肠球菌有较好活性,对屎肠球菌活性较差。
对革兰氏阴性菌,三药对流感嗜血杆菌、黏膜炎莫拉菌、淋病奈瑟氏菌具有良好活性。对于肠杆菌科细菌,左氧氟沙星和曲伐沙星的抗菌活性是三药的 2 ~ 16 倍,PGE 9262932、PGE 4175997、PGE 9509924、左氧氟沙星、曲伐沙星对环丙沙星敏感肠杆菌科细菌的 MIC90分别为 1、4、1、0.25、0.5 mg/L;对于铜绿假单胞菌、荧光假单胞菌、恶臭假单胞菌,环丙沙星和曲伐沙星的活性是三药的 2 ~ 16 倍,三药的 MIC90在 4 ~ 8 mg/L(但对环丙沙星耐药铜绿假单胞菌 MIC90> 16 mg/L)[77]。另外,它们对肺炎支原体、肺炎衣原体、军团菌属等非典型病原体也具良好活性[76]。
PGE 9262932 和 PGE 4175997 对大肠埃希菌野生型 DNA 促旋酶的 IC50值在 1.6 ~ 3.2 mg/L,与环丙沙星、曲伐沙星活性相近。对于促旋酶突变型[GyrA (Ser-83 → Trp)]菌株,这两个 6-无氟喹诺酮药物的抗菌活性是环丙沙星的 16 倍,其对该突变型促旋酶的 IC50虽升至 6.4 mg/L,但远低于环丙沙星 IC50= 102.4 mg/L[1]。PGE 9262932 和PGE 9509924 对喹诺酮耐药性决定区(quinolone resistance-determining regions,QRDRs)为野生型序列的金葡菌,抗菌活性(MIC90= 0.03 mg/L)是莫西沙星(MIC90= 0.12 mg/L)、加替沙星(MIC90= 0.25 mg/L)、左氧氟沙星(MIC90= 0.25 mg/L)、环丙沙星(MIC90= 1 mg/L)的 4 ~ 32 倍。对具有野生型 gyrA 和 parC 序列的肺炎链球菌,其活性仅受轻微影响,PGE 9262932(MIC90= 0.03 mg/L)和 PGE 9509924(MIC90= 0.12 mg/L)的抗菌活性分别是上述四个对照氟喹诺酮的 8 ~ 64 及 2 ~ 16 倍(MIC90分别为 0.25、0.5、2、2 mg/L)。对 GyrA 和 GrlA 具有双突变的金葡菌,两药的 MIC90为 4 ~ 8 mg/L,活性亦远高于上述四个对照氟喹诺酮(MIC ≥ 32 mg/L)。基于此,可以认为两药对喹诺酮耐药菌株有一定的抗菌作用。且即使细菌对青霉素、大环内酯类、复方新诺明(磺胺甲噁唑/甲氧苄啶)耐药,亦不影响它们对 PGE 9262932 的敏感性[79-80]。PGE 9509924 对肺炎链球菌的 PAE 为 3.2 ~ 10.4 h(BioScreen 法测定),长于左氧氟沙星(1.3 ~ 2.3 h)及环丙沙星(0.3 ~ 1.4 h),PGE 9262932、PGE 4175997、PGE 9509924 三者对大肠埃希菌均具有较长 PAE(2.3 ~ 2.8 h),但对铜绿假单胞菌均不具有或仅具较短 PAE(0.7 ~ 0.9 h)[81]。三药目前进一步研究进展的信息不多。

图 6 PGE 9262932结构
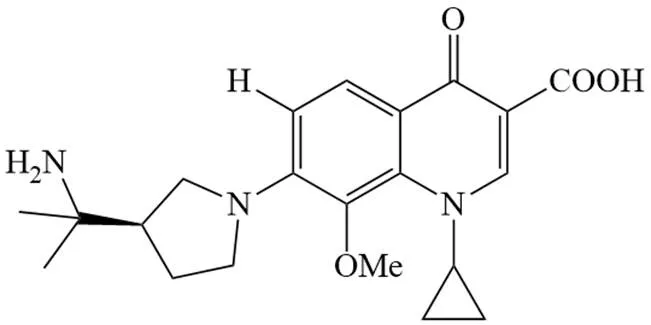
图 7 PGE 4175997结构
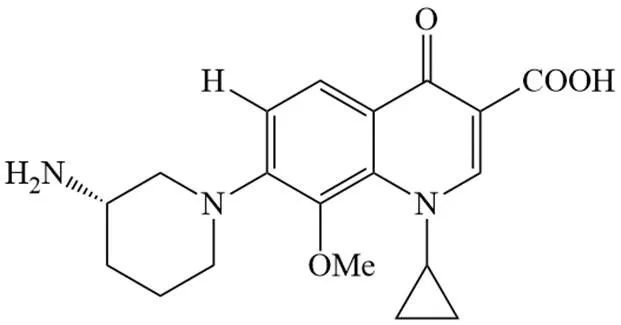
图 8 PGE 9509924结构
4 前景及展望
众所周知,氟喹诺酮类药物的成功应用是近几十年来抗菌药物研发进程中的重要里程碑,但随之而来的毒性和耐药问题也一直广受关注。系列6-无氟喹诺酮类药物的出现,在相当程度上克服了氟喹诺酮类药物应用中存在的这些问题。一方面,无氟喹诺酮类药物因 6 位去氟,遗传毒性、光毒性、过敏反应等发生率有所降低;另一方面,这类药物在保持氟喹诺酮类药物广谱抗菌活性基础上,增强了对临床氟喹诺酮耐药菌,尤其是革兰氏阳性耐药菌株的抗菌活性,为克服革兰氏阳性菌和某些革兰氏阴性菌如大肠埃希菌现有的氟喹诺酮类耐药问题提供了一种新的解决手段。此外,有研究者认为,氟喹诺酮类药物在体内代谢后平均约 25% 是以原型药形式通过排泄物、医用废水、水产饲料等途径进入环境,在水体、土壤、植物中形成不同程度富集,造成污染,破坏生态系统平衡并诱发耐药菌的产生、繁殖、传播,尤其水体中过量氟元素的存在有诱发氟斑牙等疾病的潜在风险,故发展无氟喹诺酮或能一定程度减缓氟喹诺酮应用造成的生态损害及对人体健康的危害[82]。可以认为,未来 6-无氟喹诺酮的开发与应用具有不错的前景。但总体而言,目前 6-无氟喹诺酮品种尚少,在上市药物中占比不高,比较突出的问题是临床应用数据不够充分,加之现有 6-无氟喹诺酮品种与氟喹诺酮类品种相比,对某些耐药菌和非发酵革兰氏阴性菌的体内外活性偏弱。因此,集思广益,整合现有喹诺酮药物构效关系和药化研究的最新成果,基于当代药物设计、筛选等手段,进一步优化结构,或可获得更多抗菌谱更广、药效更佳、安全性更好的新型 6-无氟喹诺酮品种,从而服务于人类的医疗健康领域。
[1] Roychoudhury S, Ledoussal B. Non-fluorinated quinolones (NFQs): new antibacterials with unique properties against quinolone-resistant gram-positive pathogens. Curr Drug Targets Infect Disord, 2002, 2(1): 51-65.
[2] Higgins PG, Fluit AC, Schmitz FJ. Fluoroquinolones: structure and target sites. Curr Drug Targets, 2003, 4(2):181-190.
[3] China Food and Drug Administration. Announcement on the revision of the instructions for systemic application of fluoroquinolones (No. 79 of 2017). (2017-07-05). http://www.nmpa.gov.cn/WS04/CL2115/ 286700.html. (in Chinese)
国家食品药品监督管理总局. 总局关于修订全身用氟喹诺酮类药品说明书的公告(2017年第79号). (2017-07-05). http://www.nmpa. gov.cn/WS04/CL2115/286700.html.
[4] U.S. Food and Drug. FDA updates warnings for fluoroquinolone antibiotics. (2016-07-26). https://www.fda.gov/news-events/press- announcements/fda-updates-warnings-fluoroquinolone-antibiotics.
[5] Sousa J, Alves G, Fortuna A, et al. Third and fourth generation fluoroquinolone antibacterials: a systematic review of safety and toxicity profiles. Curr Drug Saf, 2014, 9(2):89-105.
[6] Zhou WC,Zhang XP. New progress of fluoroquinolone research. Chin J New Drugs, 2000, 9(10):667-670. (in Chinese)
周伟澄, 张秀平. 氟喹诺酮药物研究新进展. 中国新药杂志, 2000, 9(10):667-670.
[7] Tang J, Cheng Q, Sun WX. Progress in research on non-fluorinated quinolone antibacterials. World Notes Antibiot, 2016, 37(5):198-203. (in Chinese)
唐炯, 程强, 孙文霞. 无氟喹诺酮类抗菌药研究进展. 国外医药抗生素分册, 2016, 37(5):198-203.
[8] Ledoussal B, Bouzard D, Coroneos E. Potent non-6-fluoro-substituted quinolone antibacterials: synthesis and biological activity. J Med Chem, 1992, 35(1):198-200.
[9] Fan SY, Zhou XB. Pay attention to the research progress of non-fluoroquinolone drugs. Clin Med J, 2017, 15(9):91-92. (in Chinese)
樊士勇, 周辛波. 关注无氟喹诺酮类药物研究进展. 临床药物治疗杂志, 2017, 15(9):91-92.
[10] Zhang YB, Liu ML, Guo HY. Structural characteristics of quinolone and it's structure-activity relationship to Gram-positive bacteria. World Notes Antibiot, 2008, 29(1):12-19. (in Chinese)
章怡彬, 刘明亮, 郭慧元. 新喹诺酮的结构特征及其对革兰阳性菌的构-效关系. 国外医药抗生素分册, 2008, 29(1):12-19.
[11] Liu ML, Wang JX, Guo HY. Progress of activity against mycobacteria of quinolones and it's structure-activity relationship. Chin J Pharm, 2008, 39(12):933-941. (in Chinese)
刘明亮, 王菊仙, 郭慧元. 喹诺酮抗分枝杆菌活性及其构效关系研究进展. 中国医药工业杂志, 2008, 39(12):933-941.
[12] Fàbrega A, Madurga S, Giralt E, et al. Mechanism of action of and resistance to quinolones. Microb Biotechnol, 2009, 2(1):40-61.
[13] Xu JJ, Cui L. Garenoxacin, a novel quinolone antimicrobial agent. Chin J New Drugs, 2008, 17(20):1808-1810. (in Chinese)
徐佳骏, 崔岚. 新型喹诺酮类抗菌药加雷沙星. 中国新药杂志, 2008, 17(20):1808-1810.
[14] Fung-Tomc JC, Minassian B, Kolek B, et al. Antibacterial spectrum of a novel des-fluoro(6) quinolone, BMS-284756. Antimicrob Agents Chemother, 2000, 44(12):3351-3356.
[15] Low DE, Muller M, Duncan CL, et al. Activity of BMS-284756, a novel des-fluoro(6) quinolone, against Staphylococcus aureus, including contributions of mutations to quinolone resistance. Antimicrob Agents Chemother, 2002, 46(4):1119-1121.
[16] Gradelski E, Valera L, Kolek B, et al. Comparative killing kinetics of the novel des-fluoro(6) quinolone BMS-284756, fluoroquinolones, vancomycin and beta-lactams. Int J Antimicrob Agents, 2001, 18(1): 43-48.
[17] Noviello S, Ianniello F, Leone S, et al. Comparative activity of garenoxacin and other agents by susceptibility and time-kill testing against Staphylococcus aureus, Streptococcus pyogenes and respiratory pathogens. J Antimicrob Chemother, 2003, 52(5):869-872.
[18] Goldstein EJ, Citron DM, Merriam CV, et al. In vitro activities of garenoxacin (BMS-284756) against 170 clinical isolates of nine Pasteurella species. Antimicrob Agents Chemother, 2002, 46(9): 3068-3070.
[19] Rhomberg PR, Biedenbach DJ, Jones RN. Activity of BMS284756 (T-3811) tested against anaerobic bacteria, Campylobacter jejuni, Helicobacter pylori and Legionella spp. Diagn Microbiol Infect Dis, 2001, 40(1-2):45-49.
[20] Nakatani M, Mizunaga S, Takahata M, et al. Inhibitory activity of garenoxacin against DNA gyrase of Mycoplasma pneumoniae.J Antimicrob Chemother, 2012, 67(8):1850-1852.
[21] Nagai A, Miyazaki M, Morita T, et al. Comparative articular toxicity of garenoxacin, a novel quinolone antimicrobial agent, in juvenile beagle dogs. J Toxicol Sci, 2002, 27(3):219-228.
[22] Uchida Eiji. Phase 1 clinical studies of oral garenoxacin in healthy Japanese adult subjects. Jpn J Chemother, 2007, 55(Suppl 1):95-115.
[23] Gajjar DA, Bello A, Ge Z, et al. Multiple-dose safety and pharmacokinetics of oral garenoxacin in healthy subjects. Antimicrob Agents Chemother, 2003, 47(7):2256-2263.
[24] Yamagishi Y, Hagihara M, Hamada Y, et al. Pharmacokinetic study of garenoxacin in severe renal failure patients. Jpn J Antibiot, 2015, 68(3):141-150.
[25] Aoyama T, Kamata K, Kishino S. Garenoxacin pharmacokinetics in patients undergoing maintenance hemodialysis. Hemodial Int, 2017, 21(2):206-212.
[26] Takagi H, Tanaka K, Tsuda H, et al. Clinical studies of garenoxacin. Int J Antimicrob Agents, 2008, 32(6):468-474.
[27] Wang Z, Grasela DM, Krishna G. Retrospective analysis of electrocardiographic changes after administration of oral or intravenous garenoxacin in five phase I, placebo-controlled studies in healthy volunteers. Clin Ther, 2007, 29(6):1098-1106.
[28] Kohno S, Yanagihara K, Yamamoto Y, et al. Early switch therapy from intravenous sulbactam/ampicillin to oral garenoxacin in patients with community-acquired pneumonia: a multicenter, randomized study in Japan. J Infect Chemother, 2013, 19(6):1035-1041.
[29] Miyazaki T, Nakamura S, Hashiguchi K, et al. The efficacy and safety of sitafloxacin and garenoxacin for the treatment of pneumonia in elderly patients: A randomized, multicenter, open-label trial. J Infect Chemother, 2019, 25(11):886-893.
[30] Izumikawa K, Watanabe A, Miyashita N, et al. Efficacy and safety of garenoxacin tablets on clinically diagnosed atypical pneumonia: postmarketing surveillance in Japan. J Infect Chemother, 2014, 20(9):541-548.
[31] Garg H, Katke P, Korukonda K. Post-approval surveillance on safety and clinical utility of garenoxacin mesylate in patients with community acquired respiratory tract infections. J Clin Diagn Res, 2018, 12(1):OC05-OC09.
[32] Wishart DS, Feunang YD, Guo AC, et al. DrugBank 5.0: a major update to the DrugBank database for 2018. Nucleic Acids Res, 2018, 46(D1):D1074-D1082.
[33] Vila J, Hebert AA, Torrelo A, et al. Ozenoxacin: a review of preclinical and clinical efficacy. Expert Rev Anti Infect Ther, 2019, 17(3):159-168.
[34] Sahu JK, Mishra AK. Ozenoxacin: a novel drug discovery for the treatment of impetigo. Curr Drug Discov Technol, 2019, 16(3):259-264.
[35] López Y, Tato M, Espinal P, et al. In vitro selection of mutants resistant to ozenoxacin compared with levofloxacin and ciprofloxacin in Gram-positive cocci. J Antimicrob Chemother, 2015, 70(1):57-61.
[36] Tato M, López Y, Morosini MI, et al. Characterization of variables that may influence ozenoxacin in susceptibility testing, including MIC and MBC values. Diagn Microbiol Infect Dis, 2014, 78(3):263-267.
[37] Yamakawa T, Mitsuyama J, Hayashi K. In vitro and in vivo antibacterial activity of T-3912, a novel non-fluorinated topical quinolone. J Antimicrob Chemother, 2002, 49(3):455-465.
[38] Canton R, Morrissey I, Vila J, et al. Comparative in vitro antibacterial activity of ozenoxacin against Gram-positive clinical isolates. Future Microbiol, 2018, 13:3-19.
[39] Nakajima A, Ikeda F, Kanayama S, et al. Antimicrobial activities of ozenoxacin against isolates of propionibacteria and staphylococci from Japanese patients with acne vulgaris. J Med Microbiol, 2016, 65(8):745-750.
[40] Kanayama S, Okamoto K, Ikeda Fu, et al. Bactericidal activity and post-antibiotic effect of ozenoxacin against Propionibacterium acnes.J Infect Chemother, 2017, 23(6):374-380.
[41] González Borroto JI, Awori MS, Chouinard L, et al. Studies on articular and general toxicity of orally administered ozenoxacin in juvenile rats and dogs. Future Microbiol, 2018, 13:31-40.
[42] Tarragó C, Esquirol LP, Arañó A, et al. Therapeutic efficacy of ozenoxacin in animal models of dermal infection with Staphylococcus aureus. Future Microbiol, 2018, 13(6s):21-30.
[43] Gropper S, Cepero AL, Dosik JS, et al. Cumulative irritation, sensitizing potential, phototoxicity and photoallergy of ozenoxacin in healthy adult volunteers. Future Microbiol, 2014, 9(8 Suppl):S23-S31.
[44] Gropper S, Albareda N, Santos B, et al. Systemic bioavailability, safety and tolerability of topical ozenoxacin in healthy adult volunteers. Future Microbiol, 2014, 9(8 Suppl):S11-S16.
[45] Gropper S, Albareda N, Santos B, et al. Skin tissue exposure of once- versus twice-daily topical ozenoxacin 2% cream: a Phase I study in healthy volunteers. Future Microbiol, 2014, 9(8 Suppl):S17-S22.
[46] Gropper S, Albareda N, Chelius K, et al. Ozenoxacin 1% cream in the treatment of impetigo: a multicenter, randomized, placebo- and retapamulin-controlled clinical trial. Future Microbiol, 2014, 9(9): 1013-1023.
[47] Rosen T, Albareda N, Rosenberg N, et al. Efficacy and safety of ozenoxacin cream for treatment of adult and pediatric patients with impetigo: a randomized clinical trial. JAMA Dermatol, 2018, 154(7):806-813.
[48] Wren C, Bell E, Eiland LS. Ozenoxacin: A novel topical quinolone for impetigo. Ann Pharmacother, 2018, 52(12):1233-1237.
[49] Poole RM. Nemonoxacin: First global approval. Drugs, 2014, 74(12):1445-1453.
[50] Adam HJ, Laing NM, King CR, et al. In vitro activity of nemonoxacin, a novel nonfluorinated quinolone, against 2,440 clinical isolates. Antimicrob Agents Chemother, 2009, 53(11):4915-4920.
[51] Chen YH, Liu CY, Lu JJ, et al. In vitro activity of nemonoxacin (TG-873870), a novel non-fluorinated quinolone, against clinical isolates of Staphylococcus aureus, enterococci and Streptococcus pneumoniae with various resistance phenotypes in Taiwan. J Antimicrob Chemother, 2009, 64(6):1226-1229.
[52] Lai CC, Tan CK, Lin SH, et al. Comparative in vitro activities of nemonoxacin, doripenem, tigecycline and 16 other antimicrobials against Nocardia brasiliensis, Nocardia asteroides and unusual Nocardia species. J Antimicrob Chemother, 2009, 64(1):73-78.
[53] Lauderdale TL, Shiau YR, Lai JF, et al. Comparative in vitro activities of nemonoxacin (TG-873870), a novel nonfluorinated quinolone, and other quinolones against clinical isolates. Antimicrob Agents Chemother, 2010, 54(3):1338-1342.
[54] Tan CK, Lai CC, Liao CH, et al. Comparative in vitro activities of the new quinolone nemonoxacin (TG-873870), gemifloxacin and other quinolones against clinical isolates of Mycobacterium tuberculosis.J Antimicrob Chemother, 2009, 64(2):428-429.
[55] Chotikanatis K, Kohlhoff SA, Hammerschlag MR. In vitro activity of nemonoxacin, a novel nonfluorinated quinolone antibiotic, against Chlamydia trachomatis and Chlamydia pneumoniae. Antimicrob Agents Chemother, 2014, 58(3):1800-1801.
[56] Li CR, Li Y, Li GQ, et al. In vivo antibacterial activity of nemonoxacin, a novel non-fluorinated quinolone. J Antimicrob Chemother, 2010,65(11):2411-2415.
[57] Lai CC, Lee KY, Lin SW, et al. Nemonoxacin (TG-873870) for treatment of community-acquired pneumonia. Expert Rev Anti Infect Ther, 2014, 12(4):401-417.
[58] Guo B, Wu X, Zhang Y, et al. Safety and clinical pharmacokinetics of nemonoxacin, a novel non-fluorinated quinolone, in healthy Chinese volunteers following single and multiple oral doses. Clin Drug Investig, 2012, 32(7):475-486.
[59] Cao GY, Zhang J, Zhang YY, et al. Safety, tolerability, and pharmacokinetics of intravenous nemonoxacin in healthy chinese volunteers. Antimicrob Agents Chemother, 2014, 58(10):6116-6121.
[60] Liu Y, Zhang Y, Wu J, et al. A randomized, double-blind, multicenter Phase II study comparing the efficacy and safety of oral nemonoxacin with oral levofloxacin in the treatment of community-acquired pneumonia. J Microbiol Immunol Infect, 2017, 50(6):811-820.
[61] Yuan J, Mo B, Ma Z, et al. Safety and efficacy of oral nemonoxacin versus levofloxacin in treatment of community-acquired pneumonia: a phase 3, multicenter, randomized, double-blind, double-dummy, active-controlled, non-inferiority trial. J Microbiol Immunol Infect, 2019, 52(1):35-44.
[62] van Rensburg DJ, Perng RP, Mitha IH, et al. Efficacy and safety of nemonoxacin versus levofloxacin for community-acquired pneumonia. Antimicrob Agents Chemother, 2010, 54(10):4098-4106.
[63] Chang SP, Lee HZ, Lai CC, et al. The efficacy and safety of nemonoxacin compared with levofloxacin in the treatment of community-acquired pneumonia: a systemic review and meta-analysis of randomized controlled trials. Infect Drug Resist, 2019, 12:433-438.
[64] Liu Y, Zhang Y, Zhao W, et al. Pharmacotherapy of lower respiratory tract infections in elderly-focused on antibiotics. Front Pharmacol, 2019, 10:1237.
[65] Xiao Y, Li Y, Liu J, et al. In vitro antibacterial activity of DX-619, a novel Des-F (6)-quinolone against clinical isolates in China.J Chemother, 2007, 19(6):632-642.
[66] Bogdanovich T, Esel D, Kelly LM, et al. Antistaphylococcal activity of DX-619, a new des-F(6)-quinolone, compared to those of other agents. Antimicrob Agents Chemother, 2005, 49(8):3325-3333.
[67] Wickman PA, Moland ES, Black JA, et al. In vitro activity of DX-619, a novel des-fluoro(6) quinolone, against a panel of Streptococcus pneumoniae mutants with characterized resistance mechanisms. Antimicrob Agents Chemother, 2006, 50(2):796-798.
[68] Takano T, Higuchi W, Kanda H, et al. In vitro activity of DX-619, a des-F(6)-quinolone, against Helicobacter pylori. J Antimicrob Chemother, 2011, 66(1):220-222.
[69] Tanaka K, Mikamo H, Nakao K, et al. In vitro antianaerobic activity of DX-619, a new des-fluoro(6) quinolone. Antimicrob Agents Chemother, 2006, 50(11):3908-3913.
[70] Fukuda Y, Yanagihara K, Ohno H, et al. In vivo efficacies and pharmacokinetics of DX-619, a novel des-fluoro(6) quinolone, against Streptococcus pneumoniae in a mouse lung infection model. Antimicrob Agents Chemother, 2006, 50(1):121-125.
[71] Yanagihara K, Seki M, Izumikawa K, et al. Potency of DX-619, a novel des-F(6)-quinolone, in haematogenous murine bronchopneumonia caused by methicillin-resistant and vancomycin-intermediate Staphylococcus aureus. Int J Antimicrob Agents, 2006, 28(3):212-216.
[72] Goto K, Yabe K, Suzuki T, et al. Chondrotoxicity and toxicokinetics of novel quinolone antibacterial agents DC-159a and DX-619 in juvenile rats. Toxicology, 2010, 276(2):122-127.
[73] Imamura Y, Murayama N, Okudaira N, et al. Prediction of fluoroquinolone-induced elevation in serum creatinine levels: a case of drug-endogenous substance interaction involving the inhibition of renal secretion. Clin Pharmacol Ther, 2011, 89(1):81-88.
[74] Imamura Y, Murayama N, Okudaira N, et al. Effect of the fluoroquinolone antibacterial agent DX-619 on the apparent formation and renal clearances of 6β-hydroxycortisol, an endogenous probe for CYP3A4 inhibition, in healthy subjects. Pharm Res, 2013, 30(2):447- 457.
[75] Sarapa N, Wickremasingha P, Ge N, et al. Lack of effect of DX-619, a novel des-fluoro(6)-quinolone, on glomerular filtration rate measured by serum clearance of cold iohexol. Antimicrob Agents Chemother, 2007, 51(6):1912-1917.
[76] Kim OK, Ohemeng K, Barrett JF. Advances in DNA gyrase inhibitors. Expert Opin Investig Drugs, 2001, 10(2):199-212.
[77] Barry AL, Fuchs PC, Brown SD. In vitro activities of three nonfluorinated quinolones against representative bacterial isolates. Antimicrob Agents Chemother, 2001, 45(6):1923-1927.
[78] Zhang Y. Progress in research of quinolone antibacterials. Chin J New Drugs, 2006, 15(16):1344-1350, 1356. (in Chinese)
张宇. 喹诺酮类抗菌药的研究进展. 中国新药杂志, 2006, 15(16):1344-1350, 1356.
[79] Jones ME, Critchley IA, Karlowsky JA, et al. In vitro activities of novel nonfluorinated quinolones PGE 9262932 and PGE 9509924 against clinical isolates of Staphylococcus aureus and Streptococcus pneumoniae with defined mutations in DNA gyrase and topoisomerase IV. Antimicrob Agents Chemother, 2002, 46(6):1651-1657.
[80] Adam HJ, Schurek KN, Decorby MR, et al. Comparative in vitro activity of PGE 9262932 and fluoroquinolones against Canadian clinical Streptococcus pneumoniae isolates, including molecularly characterized ciprofloxacin-resistant isolates. J Antimicrob Chemother, 2006, 58(1):202-204.
[81] Odenholt I, Löwdin E, Cars O. Postantibiotic, postantibiotic sub-MIC, and subinhibitory effects of PGE-9509924, ciprofloxacin, and levofloxacin. Antimicrob Agents Chemother, 2003, 47(10):3352-3356.
[82] He LX. Return of non-fluorinated quinolones: a novel landmark in the developing history of quinolone antimicrobial agents. Chin J Intern Med, 2020, 59(2):99-103. (in Chinese)
何礼贤. 无氟喹诺酮“回归”: 喹诺酮类药物发展史上的新标识. 中华内科杂志, 2020, 59(2):99-103.
国家自然科学基金(81273427);北京协和医学院“协和青年科研基金”(33320140177/3332016139);中国医学科学院医学与健康科技创新工程(2016-I2M-3-014);“十三五”国家科技重大专项(2018ZX 09721001)
杨信怡,Email:yangxinyi1976@hotmail.com;游雪甫,Email:xuefuyou@hotmail.com
2020-05-10
10.3969/j.issn.1673-713X.2021.01.007
