C/O杂化调变Ni催化苯分子脱氢及开环反应性能的理论研究
2021-02-04刘勇
刘 勇
(江苏省盐城技师学院 环境工程学院,江苏 盐城 224000)
焦油是一类复杂的有机混合物,含单环、多环和复杂多环芳香族化合物。焦油在低温下呈液态,极易堵塞管道、腐蚀设备、降低整体运行效率并造成环境污染,如何高效、环保、低成本地去除焦油已成为当下的研究热点。催化裂解是目前最受关注的焦油处理方式,常用的催化剂有白云石、铁矿石、橄榄石等天然催化剂,以及碱金属、铁系金属(铁、镍、钴)、碱土金属等非天然催化剂。Zhang等[1]在Ni基催化剂作用下进行了焦油催化转化实验,转化效率最高可达99%,且在12~18 h内能持续保持高转化效率,但严重的积碳和烧结导致催化剂表面孔径逐渐扩大。Wang等[2]选用萘作为焦油模型化合物,采用Ni/白云石催化剂在较低温度(700℃)下进行反应,转化效率达到95%。Anisa等[3]发现载体能极大提高催化剂的比表面积,起到增大催化剂和焦油的接触面积及抑制积碳的作用。Michel等[4]的研究结果表明,负载于橄榄石上的NiO催化剂的活性高于橄榄石单独作催化剂时的活性,且反应后Ni以Ni0及Ni-Fe合金的形式存在。
多组元Ni基催化剂有助于提高焦油的催化转化效率、降低积碳,但制备工艺复杂,成本较高。本课题组[5]提出用低品位的褐煤负载Ni基催化剂,褐煤在脱除挥发分后具有很大的比表面积,能从含Ni废水中置换出Ni离子;与Al2O3载体相比,褐煤具有更好的催化活性及抗积碳性能,但在高温下Ni与C元素发生杂化,且部分Ni被氧化形成NiO。
本工作以前期研究[5-9]为基础,对比分析了Ni、Ni/O杂化、Ni/C杂化及Ni/C/O杂化体系的电子特性及它们与苯分子之间的相互作用特性;研究了不同催化体系催化苯脱氢及开环反应的机理,揭示了褐煤负载Ni基催化剂的杂化改性对苯的作用规律,为新型Ni基催化剂的结构优化及催化反应过程控制提供理论依据,也为其他复合催化剂体系的分子设计及性能调控提供借鉴。
1 理论方法
参考Ca催化含碳物质气化的模型[10-11],基于团簇模型构建了苯分子与Ni原子、Ni/C团簇、Ni/O团簇和Ni/C/O团簇的相互作用体系,并进行几何优化得到稳定构型(如图1所示),采用该构型研究了苯分子的脱氢及开环反应。
本工作借助Materials studio计算软件,所有计算采用密度泛函理论(DFT)的GGA方法和PBE函数[11-14]。计算收敛指标:总能量2.0×10-4Ha/nm,每个原子上的力均小于0.03 eV/nm,公差偏移为1.0 nm,应力偏差为0.05 GPa,自洽精度为1.0×10-5Ha(以每个原子计)。用线性同步转变/二次同步转变方法[15]研究反应过程。
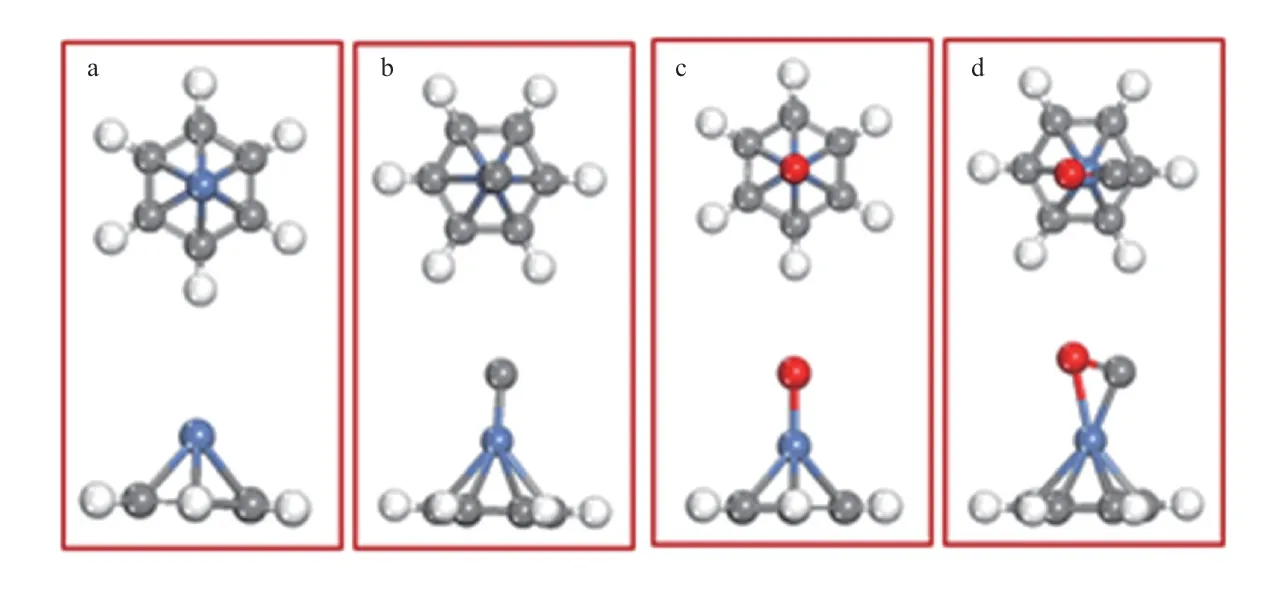
图1 苯分子与Ni原子(a)、Ni/C团簇(b)、Ni/O团簇(c)和Ni/C/O团簇(d)相互作用的稳定构型Fig.1 Stable configurations of the[benzene-Ni](a),[benzene-Ni/C](b),[benzene-Ni/O](c) and[benzene-Ni/C/O](d) interaction systems.
2 结果与讨论
2.1 苯分子与不同杂化体系的相互作用
苯分子与不同杂化体系的相互作用导致Ni原子与苯环形成稳定的η2键(如图1所示),与苯-Fe(或苯-Co)相互作用稳定构型一致[16]。相互作用的强弱可通过苯分子在表面吸附的吸附能(Ead)衡量。不同作用体系的Ead见表1。由表1可知,这些体系的Ead均为负值,即相互作用为放热过程。苯分子在各催化剂表面都能形成稳定吸附,各体系相互作用的强弱顺序为:[苯-Ni/C/O]>[苯-Ni]>[苯-Ni/O]>[苯-Ni/C]。
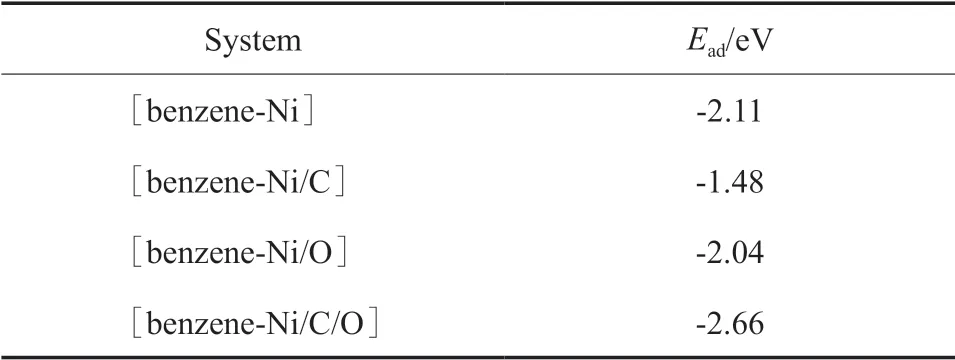
表1 不同作用体系的吸附能Table 1 Adsorption energy(Ead) for the [benzene-Ni],[benzene-Ni/C],[benzene-Ni/O]and[benzene-Ni/C/O]interaction systems
不同杂化体系下Ni原子的态密度见图2。从图2可看出,纯Ni的Ni 3d轨道电子分布在费米能级附近,容易失去电子,具有非常好的活性;当Ni/C杂化后,Ni与C原子成键,Ni 3d轨道电子向C原子转移,使得Ni的费米能级附近电子减少,同样,Ni/O杂化也使得Ni 3d轨道电子减少。而Ni/C/O杂化时,由于CO分子比较特殊,能量最高的最高占据分子轨道(HOMO)是1个δ键,在CO与Ni配位时,给出的是HOMO中的δ电子,它进入到Ni 3d轨道中,所以从图2可看出,Ni/C/O杂化使得Ni 3d轨道电子增多。
另一方面,Ni 3d轨道电子越多、越靠近费米能级,表明Ni的失电子能力越强,活性越高,因此当杂化体系与苯分子相互作用时,Ni/C/O的吸附能最高,Ni,Ni/O,Ni/C次之。从图2还可看出,Ni 3d轨道会给出电子,转移到苯分子中,与苯分子成键,导致所有体系的Ni 3d轨道上的电子减少。
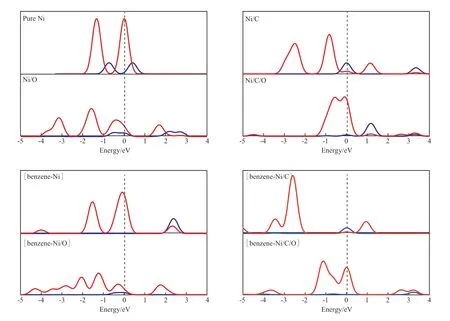
图2 不同杂化体系下Ni原子的态密度Fig.2 Partial density of state(PDOS) of Ni for different hybrid systems.
2.2 非催化条件下苯分子热解脱氢及开环反应机理
在研究苯分子热解脱氢及开环反应前,先对非催化条件下苯分子C—H键及苯环C—C键断裂反应进行过渡态搜索。图3为苯分子一步脱氢和两步脱氢生成H2分子的反应过程势能剖面。由图3可知,苯一步脱氢形成H2的反应活化能为5.05 eV,反应能为4.11 eV;而两步脱氢形成H2的过程中,第一步脱一个H原子的活化能为4.81 eV,脱出的H原子很容易与苯环上临近的H原子键合形成H2,反应活化能为0.14 eV。研究结果[17]表明,反应活化能低于0.75 eV,在温和条件下即可发生反应,所以两步脱氢形成H2的反应过程中,第一步反应是速控步。
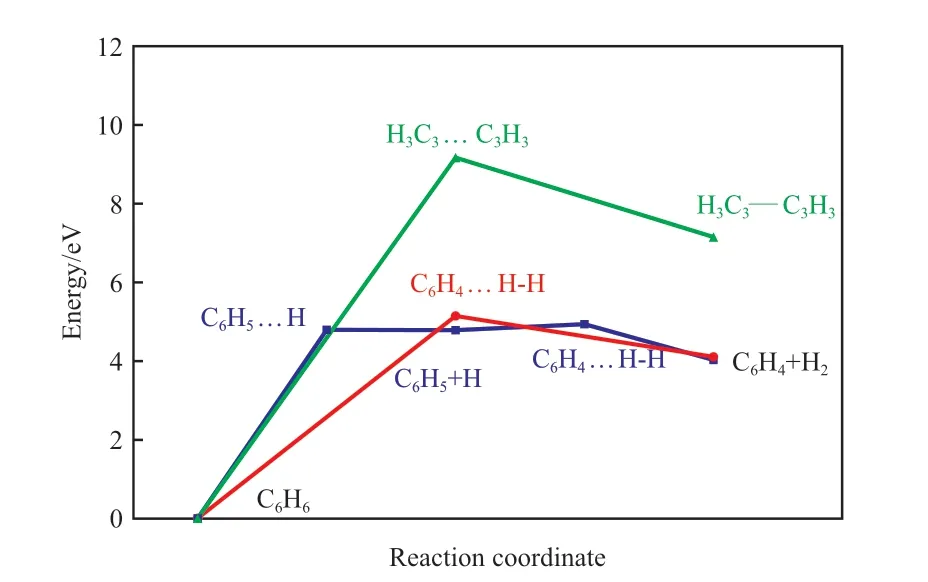
图3 苯分子脱氢及C—C键断裂开环反应过程的势能剖面Fig.3 Energy profiles for the dehydrogenation and C—C breakage reaction processes of benzene molecule.
苯的C—C键断裂开环反应的活化能为9.13 eV,反应能为7.11 eV,稍低于Muthana计算的苯环C—C键开裂反应的活化能9.63 eV[18],远高于脱氢反应所需要的活化能。苯分子在非催化条件下的脱氢反应和C—C键断裂的开环反应的活化能都远高于0.75 eV,在高温下反应仍然较难发生,这也是焦油形成的原因之一。
2.3 催化条件下苯分子热解脱氢及开环反应机理
将最稳定的[苯-Ni]、[苯-Ni/C]、[苯-Ni/O]、[苯-Ni/C/O]相互作用体系的吸附构型设为初始态,将脱一个H原子、脱一个H2分子及开环后的产物分别设为终态。通过DFT计算寻求过渡态,计算不同体系苯分子脱一个H原子、脱两个H原子形成H2及C—C键开环反应的活化能和反应能,结果见表2。不同催化体系中苯分子脱氢及C—C键断裂开环反应过程的势能剖面如图4所示。
从表2可看出,苯分子脱一个H原子时,Ni,Ni/O,Ni/C,Ni/C/O均能起到一定的催化作用,活化能和反应能都略有降低,但下降幅度较小。脱一个H2分子时,Ni和Ni/C能起到一定的催化作用,活化能和反应能都略有降低,但下降幅度较小;Ni/O和Ni/C/O会抑制催化反应,但活化能和反应能升高幅度较小,对催化反应的影响较小。催化剂催化苯分子C—C键断裂开环反应的活化能和反应能均大幅下降,远低于非催化条件下苯分子C—C键断裂反应,催化剂活性的高低顺序为:Ni>Ni/O>Ni/C>Ni/C/O。
进一步对催化开环路径上过渡态中的Ni原子和C原子的态密度进行分析(结果见图5),以了解Ni催化苯开环作用的本质。由图5可知,相比于纯苯分子,C 2p轨道态密度均发生右移,决定了催化开环反应是吸热过程。如图5a所示,过渡态C 2p轨道态密度较纯苯分子中C 2p轨道变化明显,主要是Ni与C相互作用后,Ni 3d轨道在费米能级上下发生轨道分裂,与C 2p轨道形成成键轨道和反键轨道。图5b和5c显示,苯环的C原子和Ni原子轨道均发生一定的电子重排和轨道分裂,分裂后的轨道与Ni原子轨道的重叠性不如[苯-Ni]体系(图5a)好。图5d显示C 2p轨道发生电子重排,但分裂不明显。由图5还可看出,[苯-Ni/C]、[苯-Ni/O]和[苯-Ni/C/O]的逆反应活化能低于正反应活化能。由此可见,C/O杂化在一定程度上降低了Ni基催化剂催化苯分子开环反应的转化率。
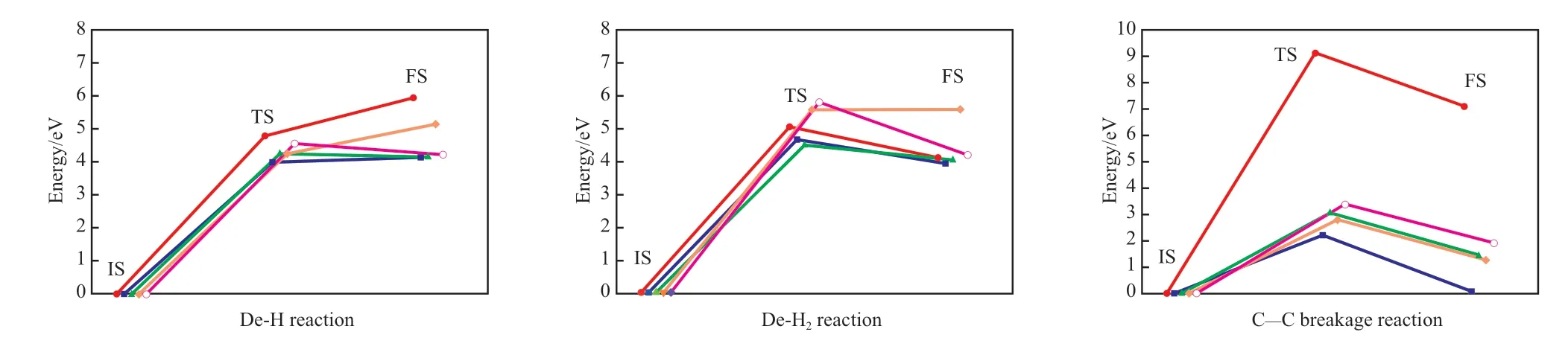
图4 不同催化体系中苯分子脱氢及C—C键断裂开环反应过程的势能剖面Fig.4 Energy profiles for the dehydrogenation and C—C breakage reactions of benzenecatalyzed by Ni,Ni/C,Ni/O and Ni/C/O.
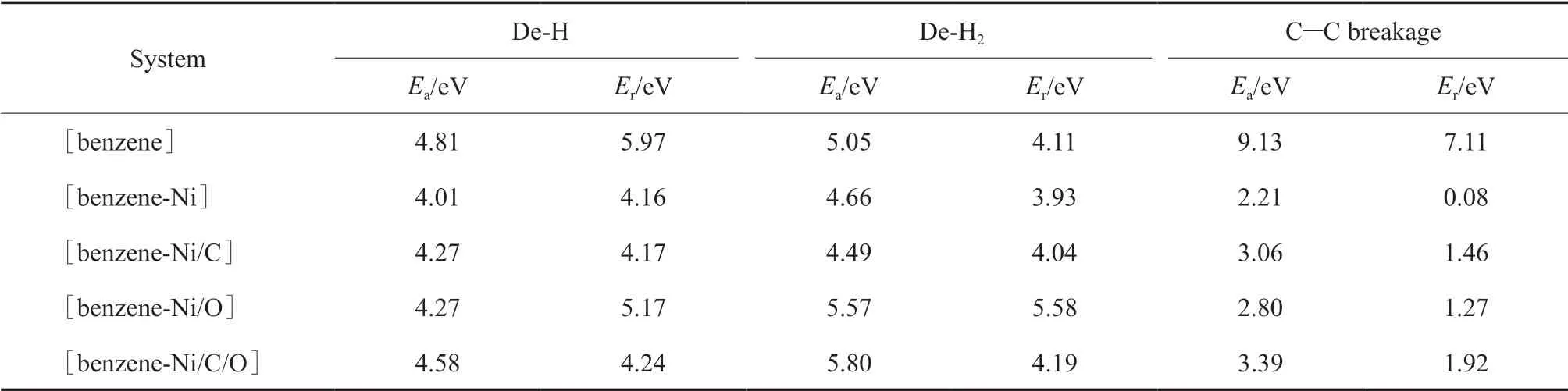
表2 苯分子脱氢及C—C键断裂开环反应的活化能和反应能Table 2 Ea and Er of dehydrogenation and C—C breakage reactions of benzene molecule
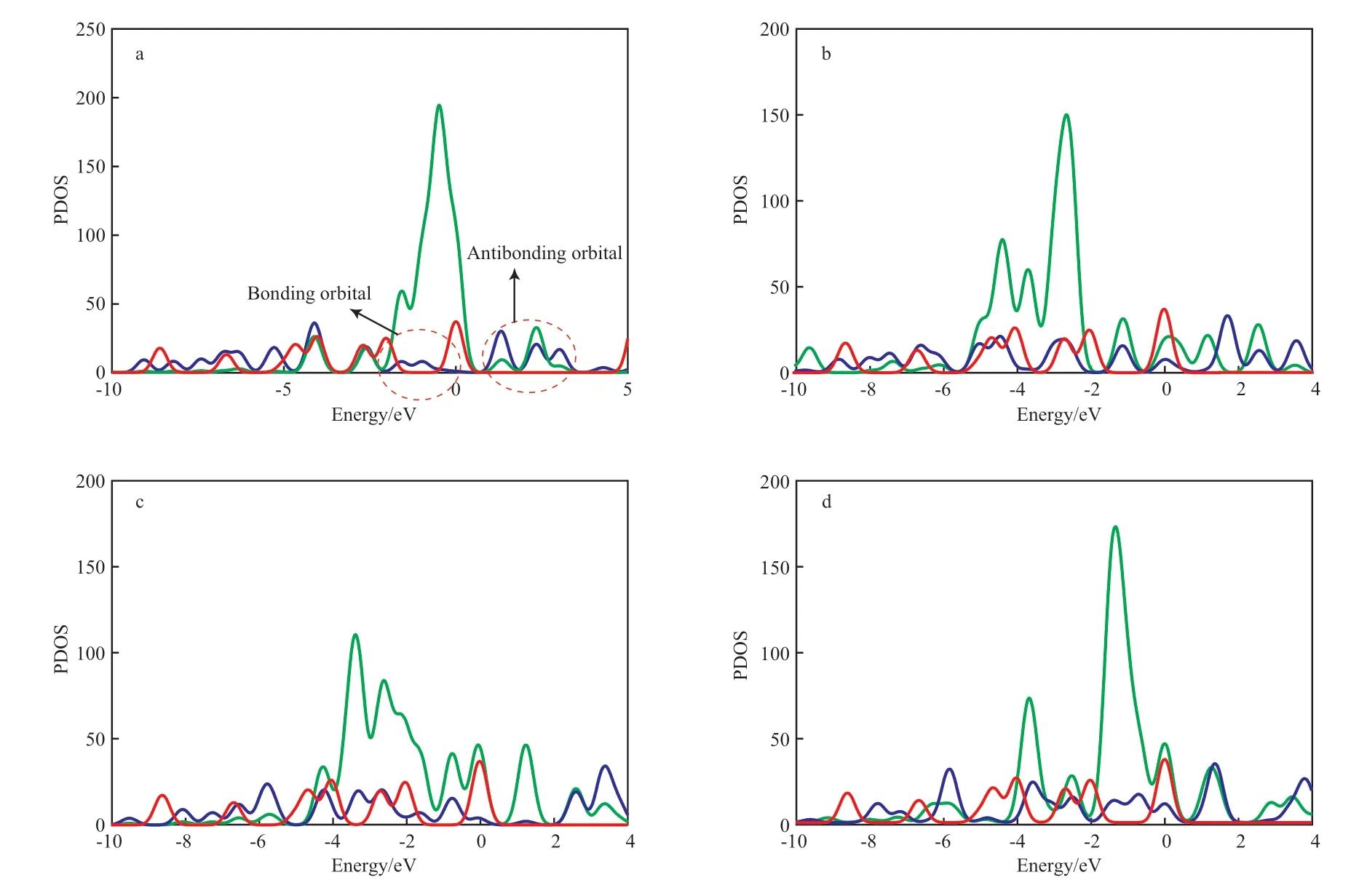
图5 催化开环反应过渡态中Ni原子和C原子的态密度Fig.5 PDOS of C and Ni atom in TS of catalytic opening ring reaction.
2.4 动力学分析
通过上述研究可以分析热力学效应对反应活化能的影响规律。已有的研究结果表明,气态分子的反应活化能与反应焓的关系符合线性Brönsted-Evans-Polanyi(BEP)关联[19-21]。苯分子C—C键断裂开环反应的反应能和活化能的关系见图6。
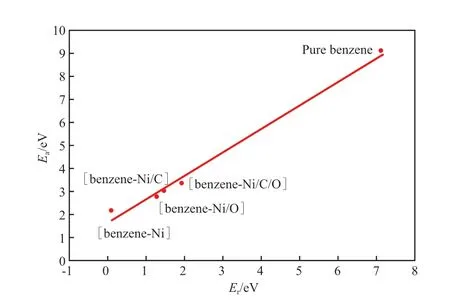
图6 苯分子C—C键断裂开环反应的反应能和活化能的关系Fig.6 Plot of Ea against Er for the C—C breakage reactions.
由图6可见,苯分子C—C键断裂开环反应的反应能与活化能符合BEP线性关系。其中,右上角高活化能区对应非催化条件下苯分子C—C键断裂反应,左下角低活化能区对应不同体系催化苯分子C—C键断裂反应。Ni、Ni/C、Ni/O和Ni/C/O均能明显降低苯分子C—C键断裂反应的活化能,使催化开环反应可在较温和的热力学条件下发生。
3 结论
1)基于DFT计算,研究了气化炉内Ni催化剂及其C/O杂化体系对焦油模型化合物苯分子的催化脱氢及开环反应机理。苯分子与Ni、Ni/O、Ni/C及Ni/C/O之间的相互作用均为放热吸附过程,各杂化体系相互作用的强弱顺序为:[苯-Ni/C/O]>[苯-Ni]>[苯-Ni/O]>[苯-Ni/C]。
2)催化剂对苯分子脱氢反应的催化作用不明显,但却能大幅降低苯分子C—C键断裂开环反应的活化能,使反应可在较温和的热力学条件下发生,催化活性顺序如下:Ni>Ni/O>Ni/C>Ni/C/O。苯分子C—C键断裂开环反应的反应能与活化能符合BEP线性关系。
3)总体上看,O杂化、C杂化,尤其是C/O同时杂化,降低了Ni基催化剂催化苯分子开环反应的转化率。在催化焦油转化反应中,应尽量控制C/O对Ni杂化导致的不利影响。多组元及多相催化剂设计也应充分考虑元素间的杂化对催化剂性能的影响。
