用于光降解罗丹明B及光解水制氢的直接Z型异质结CeO2@NiAl-LDHs:性能及机理
2021-01-29张连阳吴俊杰夏盛杰
张连阳 吴俊杰 孟 跃 夏盛杰
(1浙江省清洁染整技术研究重点实验室,绍兴文理学院纺织服装学院,绍兴 312000)(2杭州滨江房地产集团股份有限公司,杭州 310018)(3杭州信达奥体置业有限公司,杭州 310016)(4湖州师范学院生命科学学院,湖州 313000)(5浙江工业大学化学工程学院,杭州 310014)
0 引言
Z型异质结(Z-scheme heterojunction)光催化剂由于具有载流子分离效率高、氧化还原能力强的优点,近年来被广泛应用于污染物降解[1-2]、催化还原CO2[3-4]及光解水[5-6]等领域,是一种极具潜力的新型光催化剂[7-9]。Z型异质结光催化剂的能带呈交叉式排列,电子在半导体间的迁移路径类似于英文字母“Z”,因此称之为Z型异质结[10]。Wang等采用一种简便的超声方法结合光还原的策略,合成了具有高效界面电荷转移的Ag桥结构贵金属掺杂的单层氮化碳型Z型异质结光催化剂(2D/2D Bi5FeTi3O15/u1trathin g-C3N4,BFTO/Ag/UCN)[11]。通过对四环素(TC)的可见光(>420 nm)降解发现,光照 60 min 后,0.05 g BFTO/2%Ag/10%UCN(其中2%和10%均是质量分数)对20 mg·L-1的TC降解率达到了86%。但是,在此类Z型异质结中Ag或Au虽然可作为载流子转移的桥梁,但同时也加大了电子的传输距离,影响了载流子传递的稳定性,从而限制了光降解活性的进一步提高[12]。
近期有研究者提出了无介体的直接Z型异质结,其通过缩短电子的传输距离增强其稳定性[13]。Zhao等制备了新型三维微球无介体Z型Ag3VO4/Bi-VO4异质结光催化剂[14]。该催化剂表现出了高效的光催化还原Cr6+(74.9%)和光降解双酚S(94.8%)活性。该Z型异质结中并没有其他物质作为载流子转移的桥梁,而是Ag3VO4和BiVO4形成无介体的直接Z型异质结,大大缩短了载流子的传递路径,增强了传递的稳定性。Zhang等在一维空心Co9S8纳米管上生长了2D结构的ZnIn2S4纳米片,制备了Co9S8/ZnIn2S4多层管状的Z型异质结[15]。实验结果表明,Co9S8/ZnIn2S4光催化剂具有优异的光催化活性,可在45 min内将Cr(Ⅵ)100%还原,其最高析氢速率为90.3 mg·g-1·h-1。在不使用任何助催化剂的情况下,Co9S8/ZnIn2S4的产氢速率比纯ZnIn2S4高11.2倍。作者认为,高催化活性的原因是所构建的Co9S8/ZnIn2S4多层中空结构具有良好的光吸收能力、较大的比表面积和丰富的催化活性位点。此外,光/电化学表征表明,Co9S8的引入可以显著提高光生电子和空穴的分离和转移效率。然而,要基于Z型异质结设计和研制新型光催化材料,实现可见光降解高浓度的有机污染物或分解水制氢,在活性与光催化机理方面仍存在很大的问题[16-17]。
由于核壳结构催化剂具有大比表面积,通常可暴露更多的表面活性位;同时,壳体通过表面包覆形成特殊的梯度结构可以将外壳粒子特有的光学性能和催化性能赋予内核粒子[18-19]。因此,我们基于典型的阴离子型层状黏土矿物水滑石(1ayered doub1e hydroxides,LDHs)[20-22],拟利用金属氧化物@LDHs纳米复合物构筑核壳结构的Z型异质结光催化剂。将其应用于高效的可见光降解有机污染物和分解水制氢。结合材料表征和密度泛函理论(DFT)计算,分析异质结内部内建电场的形成过程和加速光生电子转移的机制,探讨光生电子传递的方向和数量,给出光催化机理。
1 实验部分
1.1 CeO2@NiAl-LDHs异质结的制备
1.1.1 CeO2的合成
以 CeC13·7H2O、NH3·H2O、十 二 烷 基 硫 酸 钠(SDS)作为原料,采用反向沉淀法制备纳米CeO2。在室温下,将一定量的SDS和CeC13·7H2O溶于60 mL去离子水(Ce4+和SDS的物质的量之比为2∶1)。之后向溶液中加入一定量的氨水,保持溶液的pH值为8.5,而后以300 r·min-1的搅拌速度反应2 h。生成的沉淀物经离心和去离子水洗涤各3次,然后在80℃下干燥24 h。干燥后的样品放入管式炉中在300℃温度下煅烧2 h,得到纳米CeO2样品。
1.1.2 NiA1-LDHs的合成
水滑石典型的合成过程如下:在氮气保护下,将 0.15 mo1(43.65 g)Ni(NO3)2·6H2O 和 0.05 mo1(18.75 g)A1(NO3)3·9H2O溶于100 mL去离子水中配成溶液A,将0.36 mo1(14.40 g)NaOH溶于100 mL去离子水中配成溶液B。室温下,以300 r·min-1的搅拌速度,将溶液A和B同时滴加于含有50 mL去离子水的三口烧瓶中,控制溶液pH值为8.5~9.0,整个滴加过程控制在30 min。所得的浆液在65℃下晶化24 h,而后经离心、洗涤至中性,再将样品在85℃下真空干燥12 h,所得样品为NiA1-NO3-LDHs,记为NiA1-LDHs。
1.1.3 CeO2@NiA1-LDHs的合成
实验中所用的水均为去CO2的重蒸水(将去离子水煮沸去除约1/5的体积)。将一定质量的CeO2(5、10、15mg)、3mmo1Ni(NO3)2·6H2O、1mmo1A1(NO3)3·6H2O、4 mmo1 NH4F和10 mmo1尿素溶于20 mL的去离子水中,超声15 min,再搅拌30 min使反应体系分散均匀。将上述溶液转移至Tef1on反应釜中(事先用氮气尽可能地排出釜内空气),在100℃的烘箱中反应12 h。反应结束后取出反应釜,自然冷却至室温。将上述溶液进行离心、洗涤,用100 mL去离子水和100 mL无水乙醇各洗涤3次。经50℃真空干燥12 h后得到的产物为CeO2@NiA1-LDHs-1、CeO2@NiA1-LDHs-2、CeO2@NiA1-LDHs-3(1、2、3分别代表CeO2质量为5、10、15 mg)。
1.2 材料表征
在Rigaku U1timaIV粉末衍射仪上进行粉末X射线衍射(XRD)测试,电压为40 kV,电流为178 mA,CuKα辐射(λ=0.154 nm),扫描速率为5(°)·min-1,扫描范围为5°~70°。C、H、N和O元素的微量分析在ThermoFisher Ita1ia S.P.A元素分析仪上进行;在IRIS IntrepidⅡXSP仪器上使用电感耦合等离子体原子发射光谱法(ICP-AES)测定Ce、Ni和A1元素的含量。扫描电镜-能量散射X射线谱(SEM-EDS)分析在日立SU1510 ESEM中进行,加速电压为15 kV。采用日立HT-7700透射电镜(TEM,200 kV)表征样品的形貌、晶格条纹和晶界。用Micromeritics Instrument Corporation ASAP2020M仪器,在77 K下通过N2吸附-脱附分析材料的孔结构。测试之前,将样品在120℃的真空中脱气6 h。通过Brunauer-Emmett-Te11er(BET)方法计算比表面积。采用Brunauer-Joyner-Ha11enda(BJH)法测定了样品的孔径分布和总孔容。用岛津UV-2600型紫外光谱仪在室温下记录了空气中的固态紫外可见漫反射光谱,以固体BaSO4为背景。基于锁定的表面光电压(SPV)光谱测量系统由单色光源、样品池、计算机和带光斩波器(SR540)的锁定放大器(SR830-D SP)组成,采用24 Hz的低斩波频率,500 W氙灯(CHF-XM-500 W),光栅单色仪(Omni-5007,Zo1ix)提供单色光。
1.3 光催化降解罗丹明B
采用可调节入射光强度(200~600 W)的氙灯作为模拟光源,采用滤光片滤去λ<400 nm和λ>800 nm的干扰光。以一定浓度的罗丹明B(RhB,30~60 mg·L-1)为目标降解物来测试催化剂的光催化性能。实验中将10~30 mg催化剂加入盛有50 mL RhB溶液的双层石英反应管中。首先在避光条件下搅拌30 min,以达到材料对RhB的吸附-脱附平衡。随后打开模拟光源(石英管与光源距离为15 cm),在持续的光照和磁力搅拌下进行光降解实验。根据反应条件的差异,实验过程中用0.1 mo1·L-1的盐酸和氢氧化钠控制体系的pH值为3~11,反应温度为15~55℃。每隔一段时间(20~60 min)从石英反应管中取2 mL溶液,离心后取上清液,通过紫外可见分光光度计(552 nm)测定吸光度,最终计算其降解率R=(c0-ct)/c0(其中,ct为降解t时刻时 RhB的浓度,c0为暗吸附后RhB的初始浓度)。
1.4 光催化分解水制氢
以150 W的中压汞灯为可见光源,采用滤光片滤去λ<400 nm和λ>800 nm的干扰光,进行光催化分解水制H2的实验。该可见光源的光照强度约为100 mW·cm-2。进行制氢反应时,将50 mg催化剂分散在30 mL体积分数为10%的CH3OH水溶液中作为牺牲试剂。在反应之前,用氮气吹扫反应器以保持惰性气氛,然后不断搅拌催化剂悬浮液直到反应结束。采用具有0.5 nm分子筛填充柱的气相色谱(GC-16A)检测生成的H2,检测器为热导检测器(TCD)。
2 结果与讨论
2.1 核壳结构CeO2@NiAl-LDHs催化剂的表征
图1A是样品的XRD图。从中可以看出,CeO2样品出现了(111)、(200)、(220)和(311)的晶面衍射峰,与CeO2的标准PDF卡片(PDF No.34-0394)完全一致[23]。NiA1-LDHs的XRD图包含了典型的水滑石样品 的 (003)、(006)、(009)/(012)、(015)、(018)、(110)和(113)的晶面衍射峰,峰型完好且尖锐[24-26]。而CeO2@NiA1-LDHs-2的XRD图则完整地包含了上述2个组分的所有特征衍射峰。此外,样品的元素分析结果列于表1,测试结果与预计的样品组成与含量基本一致。上述结果充分表明,成功制备了CeO2@NiA1-LDHs复合物。
样品的N2吸附-脱附等温线以及由BET方法计算得到的比表面积数据如图1B和表1所示。由图可知,CeO2和NiA1-LDHs的比表面积仅为38和55 m2·g-1,由于形成了核壳结构使得CeO2@NiA1-LDHs-2的比表面积增大为96 m2·g-1。
图1C是样品的紫外可见漫反射光谱图。其中,CeO2在可见光区的吸收较少,NiA1-LDHs和CeO2@NiA1-LDHs-2在400~800 nm有明显的可见光吸收。CeO2、NiA1-LDHs和CeO2@NiA1-LDHs-2的禁带宽度分别为2.92、2.53和2.69 eV(图1D)。

图1 CeO2、NiA1-LDHs和CeO2@NiA1-LDHs-2的XRD图 (A)、N2吸附-脱附等温线 (B)、紫外可见漫反射谱图(C)和禁带宽度(D)Fig.1 XRD patterns(A),N2adsorption-desorption isotherms(B),u1travio1et-visib1e diffuse ref1ectance spectra(C)and band gaps(D)of CeO2,NiA1-LDHs and CeO2@NiA1-LDHs-2
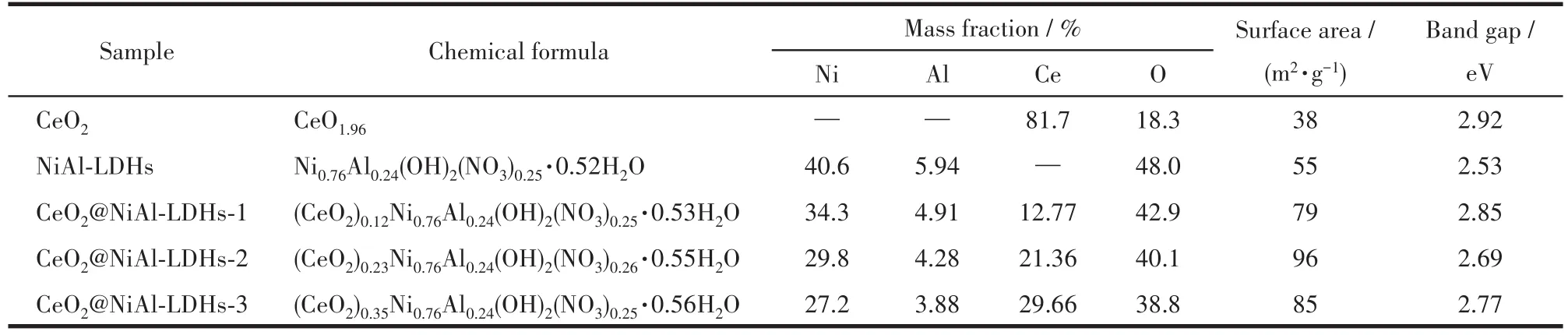
表1 样品的化学式、元素含量、比表面积和禁带宽度Table 1 Chemical formula,element contents,surface area and band gap of the samples
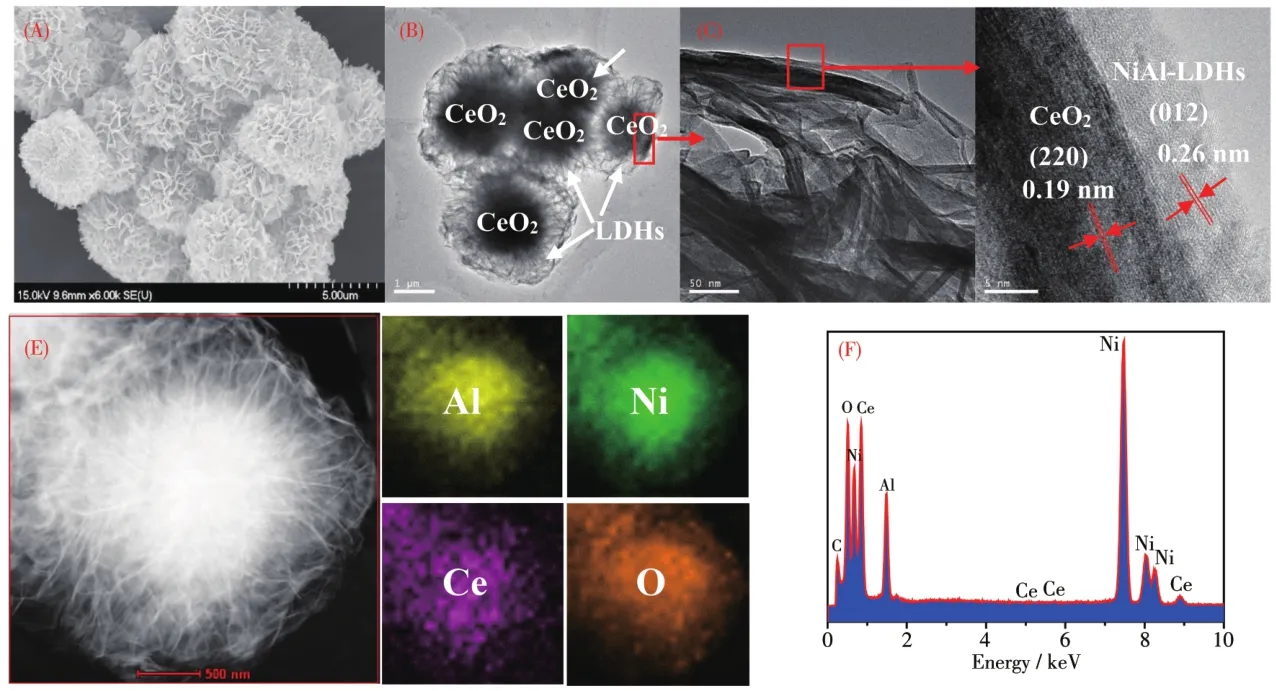
图2 CeO2@NiA1-LDHs-2的SEM图(A)和TEM图(B~D)、HAADF-STEM图以及相应的元素mapping图(E)和EDS谱图(F)Fig.2 SEM image(A)and TEM images(B~D),HAADF-STEM image,corresponding e1ementa1 mappings(E)and EDS spectrum(F)for CeO2@NiA1-LDHs-2
CeO2@NiA1-LDHs-2样品的SEM图和TEM图如图2所示。从SEM图可以看出,CeO2@NiA1-LDHs-2呈球形的簇状结构,且尺寸均一、规整(图2A)。图2B~2D是CeO2@NiA1-LDHs-2样品在不同倍数下的TEM和HRTEM图。从中可以清楚地看到代表NiA1-LDHs的外层间距为0.26 nm的(012)晶面以及内层CeO2的(220)晶面(0.19 nm)。上述结果充分说明,合成的样品具有球形的核壳结构,其以CeO2为内核,NiA1-LDHs纳米片为外壳。图2E是CeO2@NiA1-LDHs-2样品的高角度环形暗场-扫描透射电子显微镜(HAADF-STEM)和元素mapping数据。结果表明,样品含有明显的Ce、A1、Ni和O等元素。另外,EDS表明(图2F),上述元素的组成与ICP-AES测试得到的结果(表1)基本一致,这也进一步说明CeO2@NiA1-LDHs具有核壳结构。
2.2 可见光降解RhB和光解水制氢的性能
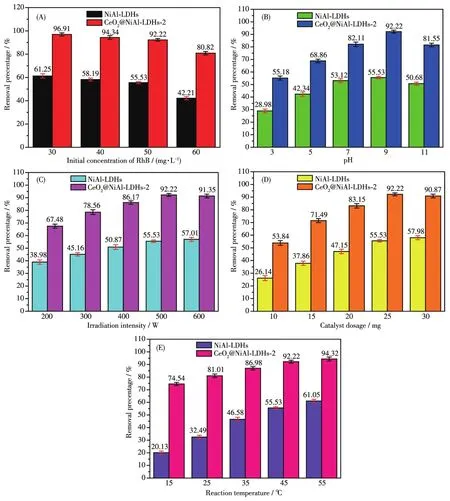
图3 RhB的去除率与起始浓度(A)、pH值(B)、光照强度(C)、催化剂用量(D)和反应温度(E)的关系Fig.3 Re1ationship between remova1 percentage of RhB and initia1 concentration(A),pH(B),irradiation intensity(C),cata1yst dosage(D)and reaction temperatures(E)
我们选择了典型的偶氮染料RhB作为目标降解物,测试了材料的光催化氧化性能。光催化实验前,均在黑暗处进行了30 min的吸附实验,结果表明,NiA1-LDHs、CeO2@NiA1-LDHs-2对RhB的吸附率均不大于12%。因此,后续的光催化实验均未扣除吸附的影响。而后,我们进行了光催化反应条件的优化,测试的条件参数和范围:RhB起始浓度为30~60 mg·L-1,pH=3~11,光照强度为 200~600 W,催化剂用量为10~30 mg,反应温度为15~55℃,结果列于图3A~3E。以CeO2@NiA1-LDHs-2为例,除了pH值的影响呈现一个先上升后下降的趋势(在pH=9时活性最大)外,其他条件下均呈现出较好的线性关系。虽然RhB的去除率随着各影响因素数值的增大呈线性地提高或减小,但可以明显地看出当起始浓度超过50 mg·L-1、光照强度超过500 W、催化剂用量超过25 mg、反应温度大于45℃时,去除率的提高效果明显减小(或降低幅度很大)。因此,考虑到综合效率,我们认为最佳的反应条件:RhB的起始浓度为50 mg·L-1,pH=9,光照强度为500 W,催化剂用量为25 mg,反应温度为45℃,此时去除率为92.2%。此外,我们对影响NiA1-LDHs和CeO2@NiA1-LDHs-2光降解RhB的因素,包括pH值、光照强度、催化剂用量和反应温度进行了动力学拟合,结果列于图4。所有数据较好地符合准一级动力学方程,相关系数均大于0.98;同时CeO2@NiA1-LDHs-2的反应速率常数均明显大于NiA1-LDHs,与活性结果相吻合。
在最佳条件下,催化剂光降解RhB的浓度随时间的变化曲线如图5A所示。由图可知,反应在300 min达到平衡时,CeO2和NiA1-LDHs的RhB去除率仅为24.1%和55.5%,而CeO2@NiA1-LDHs则最高达92.2%(CeO2@NiA1-LDHs-2)。图5B是催化剂降解RhB的活性数据,CeO2和NiA1-LDHs的活性仅为9.65和22.33 mg·g-1·h-1,而CeO2@NiA1-LDHs-2的活性最高可达36.91 mg·g-1·h-1。同时我们也对样品进行了光催化分解水制氢测试,结果列于图5C和5D中。与氧化降解的结果类似,CeO2和NiA1-LDHs的活性仅为 1.90 和 5.52 mmo1·g-1·h-1,而 CeO2@NiA1-LDHs制氢的活性最高可达14.08 mmo1·g-1·h-1(CeO2@NiA1-LDHs-2),活性提高得非常明显。上述氧化降解有机污染物和分解水制氢的实验结果表明,CeO2@NiA1-LDHs复合物可大幅提升CeO2和NiA1-LDHs的光催化性能。
此外,我们还测试了CeO2@NiA1-LDHs-2样品的光催化重复利用性能,结果列于图5E和5F。经过5次的循环利用,材料对RhB的光催化降解去除率从原先的92.2%仅仅下降到82.2%,而制氢性能由70.4 mmo1·g-1降低到 66.4 mmo1·g-1。上述结果充分表明材料具有稳定的光催化性能和重复利用性能。
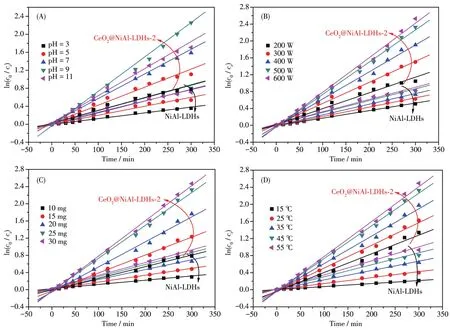
图4 NiA1-LDHs和CeO2@NiA1-LDHs-2光降解RhB的动力学拟合:pH值(A)、光照强度(B)、催化剂用量(C)和反应温度(D)Fig.4 Kinetics fitting resu1ts of NiA1-LDHs and CeO2@NiA1-LDHs-2 for photodegradation of RhB:pH(A),irradiation intensity(B),cata1yst dosage(C)and reaction temperature(D)
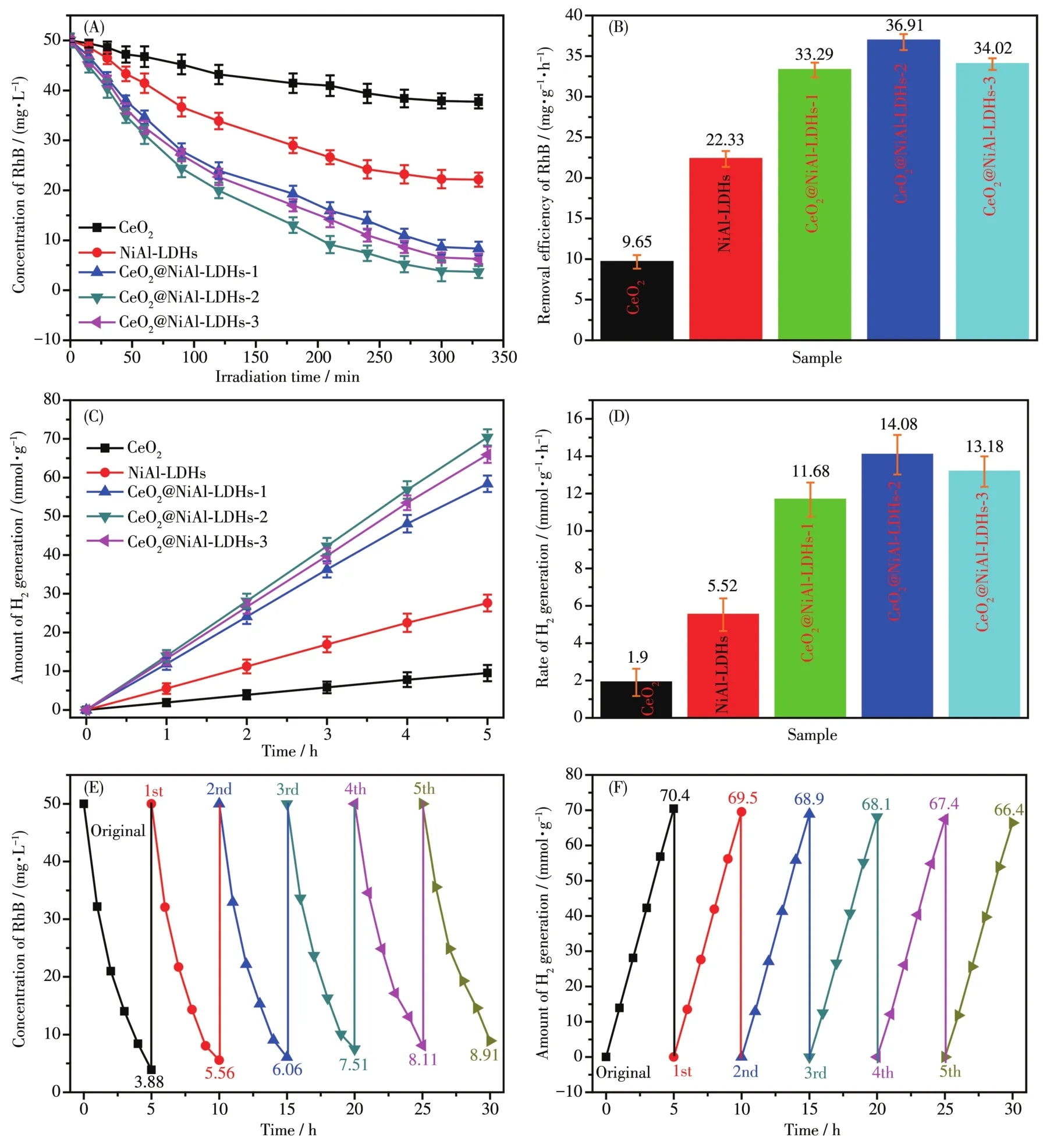
图5 不同的催化剂降解RhB的浓度随时间的变化曲线(A)和去除效率(B);氢气的产率随时间的变化曲线(C)和产氢活性(D);CeO2@NiA1-LDHs-2光降解(E)和光解水制氢(F)的重复利用结果Fig.5 Re1ationship between concentration of RhB and irradiation time(A)and remova1 efficiency of RhB(B)photodegraded by different samp1es;Re1ationship between amount of H2generation and irradiation time(C),and rate of H2generation(D);Recyc1ing resu1ts of RhB photodegradation(E)and hydrogen evo1ution(F)of CeO2@NiA1-LDHs-2
2.3 光催化机理
图6A是CeO2@NiA1-LDHs的结构模型示意图,用于后续的理论计算[27-28]。图6B是CeO2和NiA1-LDHs的价带-光电子能谱(VB-XPS)曲线,从中可以计算出两者的VB分别为2.64和1.56 eV。样品的禁带宽度数据由紫外曲线计算得到(表1),基于公式ECB=EVB-Eg可知(ECB为导带,EVB为价带,Eg为禁带宽度)[29],CeO2和 NiA1-LDHs的 CB 分别为-0.28和-0.97 eV。
当两相紧密接触形成异质结时,由于材料的费米能级不同,两者会发生电荷转移,直至费米能级趋于一致,此过程为电子重排过程。电子重排过程中遵循高费米能级→低费米能级,但是费米能级很难直接测量。因此我们引入了功函数(Φ)的概念。根据文献可知EF=Evac-Φ[30],其中EF、Φ和Evac分别代表费米能级、功函数和真空能级(Evac是处于真空水平的静止电子的能量,假定为0)。图6C和6D是由DFT计算所得的Φ的结果。CeO2和NiA1-LDHs的Φ分别为4.35和3.52 eV。因此,两者的EF则为-4.35和-3.52 eV,表明当两相接触构成异质结时,电荷不断地从NiA1-LDHs向CeO2发生转移,直至两者的EF趋于一致(图7A和7B)。电子发生重排后,形成的异质结两相间存在电势差,从而形成内建电场。NiA1-LDHs由于失去电荷成为正极,CeO2得到电荷成为负极,这样构成一个从NiA1-LDHs到CeO2的内建电场(图7B)。由之前的分析可知,CeO2@NiA1-LDHs异质结内部的两相,即CeO2和NiA1-LDHs均可在可见光激发下产生载流子,也就是光生电子和空穴。载流子由于带电荷,会在内建电场的诱导作用下在两相之间进行传递。以电子为例,由于电场方向为NiA1-LDHs到CeO2,光生电子的传递方向则相反,为CeO2到NiA1-LDHs。
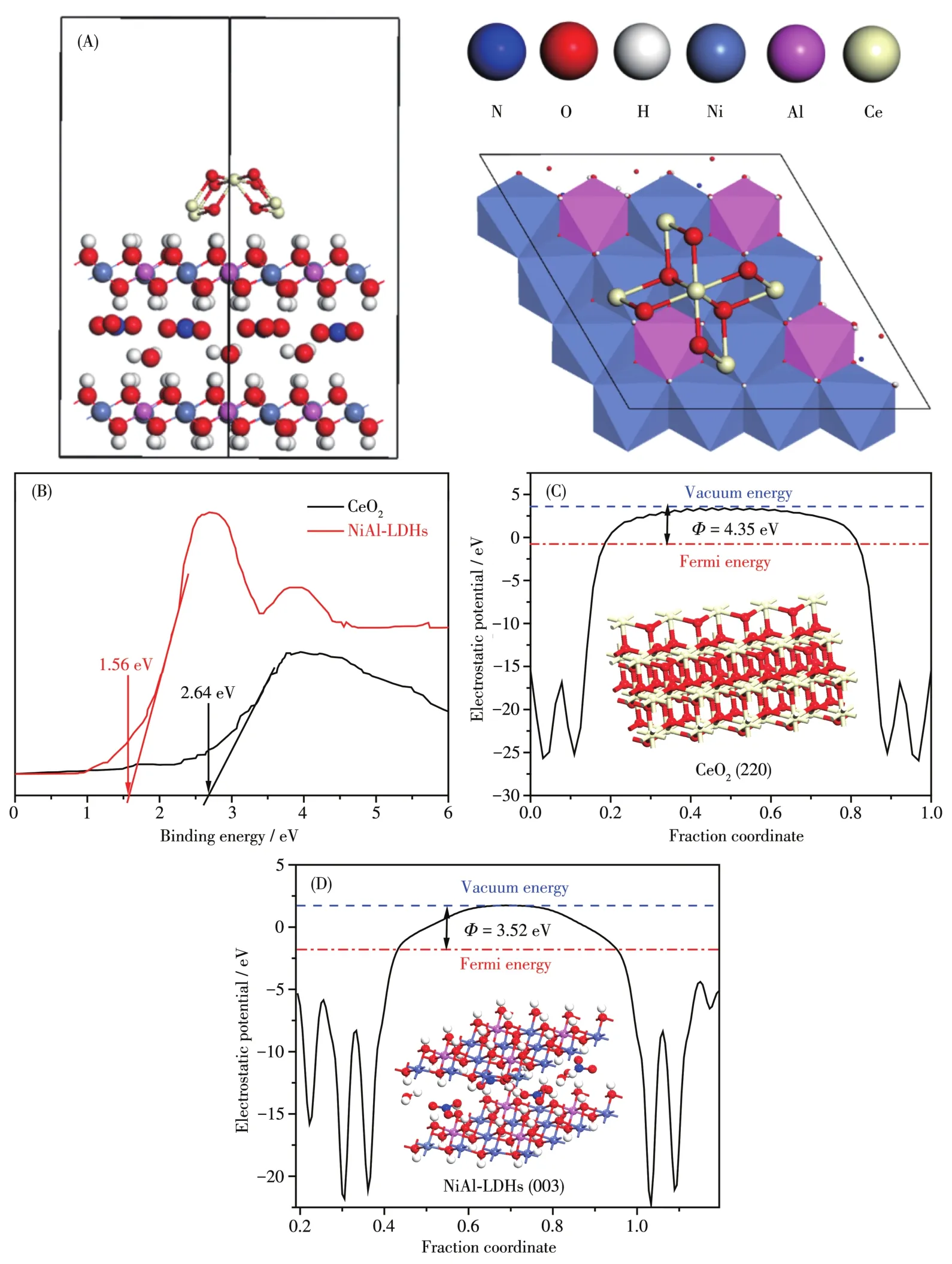
图6 (A)CeO2@NiA1-LDHs的结构模型:正视图(左),俯视图(右);(B)CeO2和NiA1-LDHs的VB-XPS测试;(C)CeO2和(D)NiA1-LDHs的功函数计算结果(电子势能图)Fig.6 (A)Structura1 mode1s of CeO2@NiA1-LDHs:front view(1eft),top view(right);(B)VB-XPS spectra of the CeO2and NiA1-LDHs;Ca1cu1ated working function(e1ectrostatic potentia1s diagrams)for(C)CeO2and(D)NiA1-LDHs
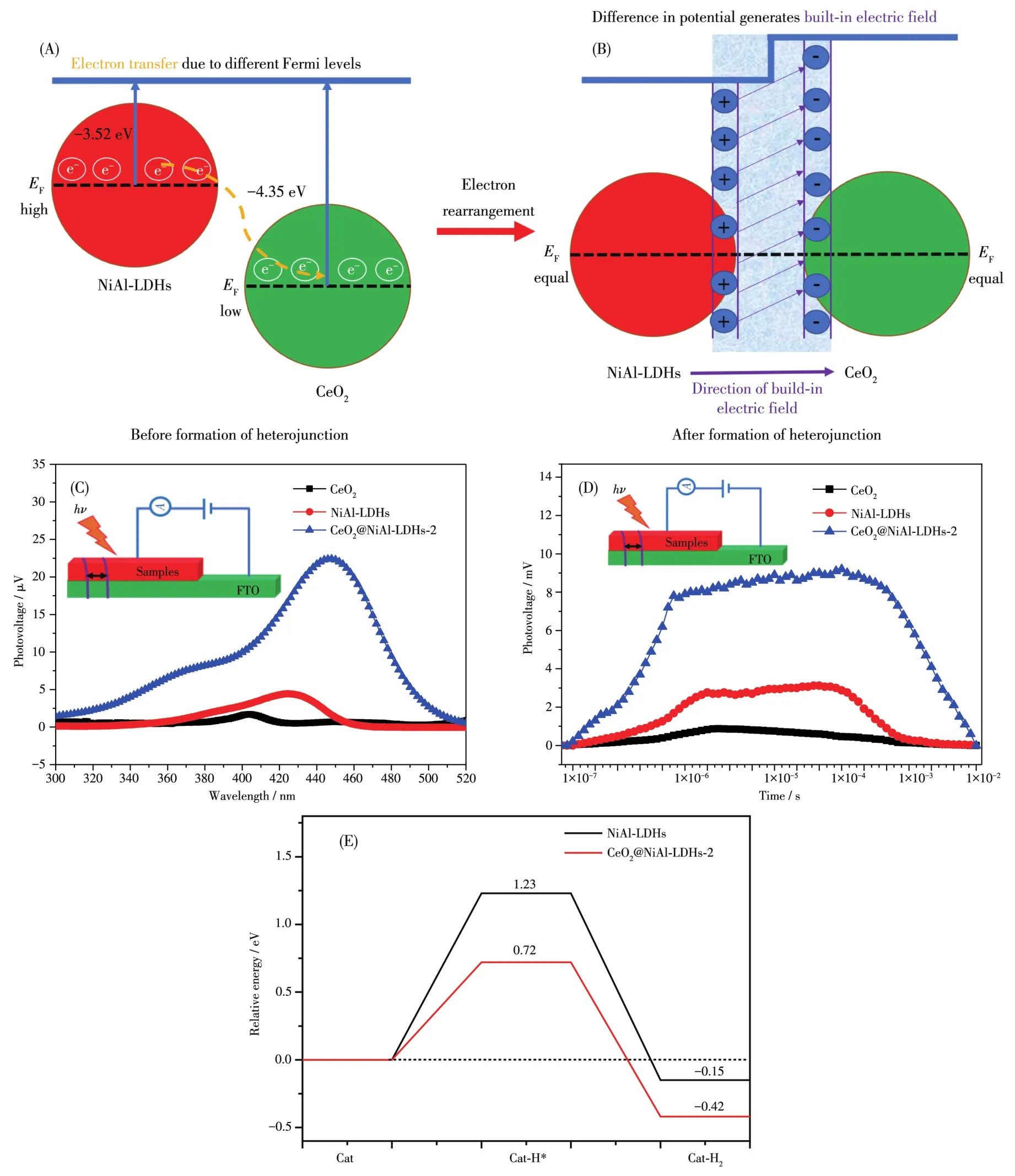
图7 CeO2@NiA1-LDHs异质结内部电荷转移(A)和内建电场的形成(B)示意图;CeO2、NiA1-LDHs和CeO2@NiA1-LDHs-2的SPV(C)、TPV(D)和基于DFT计算的产氢过程吸附能变化(E)Fig.7 Schematic for the charge-transfer process(A)and formation of bui1d-in e1ectric fie1d(B)in CeO2@NiA1-LDHs heterojunction;SPV(C)and TPV(D),ca1cu1ated adsorption energy profi1es during hydrogen production(E)for CeO2,NiA1-LDHs and CeO2@NiA1-LDHs-2
为了证实这种由于内建电场的构建而加速的电子在异质结内部的传递,我们对3种材料进行了SPV和瞬态光电压(TPV)的测试。图7C是3个样品的SPV测试结果。一般来说,当光生电子和空穴在材料内部被分离时,SPV响应信号开始上升,电子转移越多,信号强度越强[31]。CeO2在整个波长区域内SPV响应信号基本可忽略。由于NiA1-LDHs存在固有跃迁,其在350~450 nm的区域内有明显的信号峰但强度不高,说明光生载流子在此区域可实现较弱的分离。而CeO2@NiA1-LDHs在360~520 nm范围内具有明显的SPV响应信号强度,其峰强(顶峰数值)是CeO2和NiA1-LDHs的20倍和5倍。这充分说明,相对于CeO2和NiA1-LDHs,异质结内部光生电子和空穴转移的强度明显增大(数量增多)。这有力地证明了CeO2和NiA1-LDHs构成异质结后,内部形成了内建电场,加速了电子和空穴的分离。
图7D为3个样品的TPV测试结果。通常,TPV数据可提供光生电荷转移的动力学信息:(1)时间刻度小于10-5s的快速部分是光生电子和空穴在内建电场作用下的分离过程;(2)时间刻度大于10-4s的慢速部分则为光生载流子的扩散过程[32]。CeO2和NiA1-LDHs的TPV曲线均只有第一个阶段(快速部分),且信号强度均不大,说明2种材料内部价电子的传递作用很小,而光生电子和空穴的复合作用很强。CeO2@NiA1-LDHs-2的TPV信号在极短的时间内强度快递增加(快速部分),说明了光生电子和空穴的快速分离;同时在慢速部分,信号的跨度和持续时间明显延长,这说明光生电子和空穴的复合被有效地抑制,使得光生载流子的寿命得到了极大的延长。
另外,第一性原理的计算也表明,CeO2@NiA1-LDHs的H*形成和H2解吸的能垒从NiA1-LDHs的1.12和0.67 eV明显降低到-0.11和-0.39 eV。这也进一步证明了光催化产氢反应在异质结表面反应动力学的加速(图7E)。
根据以上分析,我们提出了Z型异质结CeO2@NiA1-LDHs的光催化机理(图 8):(1)由于CeO2和NiA1-LDHs的禁带宽度分别为2.92和2.53 eV,因此在可见光激发下,两者都可以产生光生载流子,即VB电子转移到CB形成光生电子,VB上形成光生空穴。(2)异质结在两相之间形成,由于费米能级的差异,电子在两相间发生迁移,异质结内部形成内建电场。在内建电场的诱导下,CeO2的CB中的电子向NiA1-LDHs的VB发生转移(其中少部分会与VB上的空穴进行复合),这样就使得具有高还原性的光生电子和高氧化性的空穴分别在NiA1-LDHs的CB和CeO2的VB积累,从而实现载流子的高效分离。(3)具有强还原性的光生电子一部分直接用于氢的还原,一部分用来捕获水中的O2生成·O2-从而氧化RhB;而空穴捕获H2O分子生成·OH,用于RhB的降解。
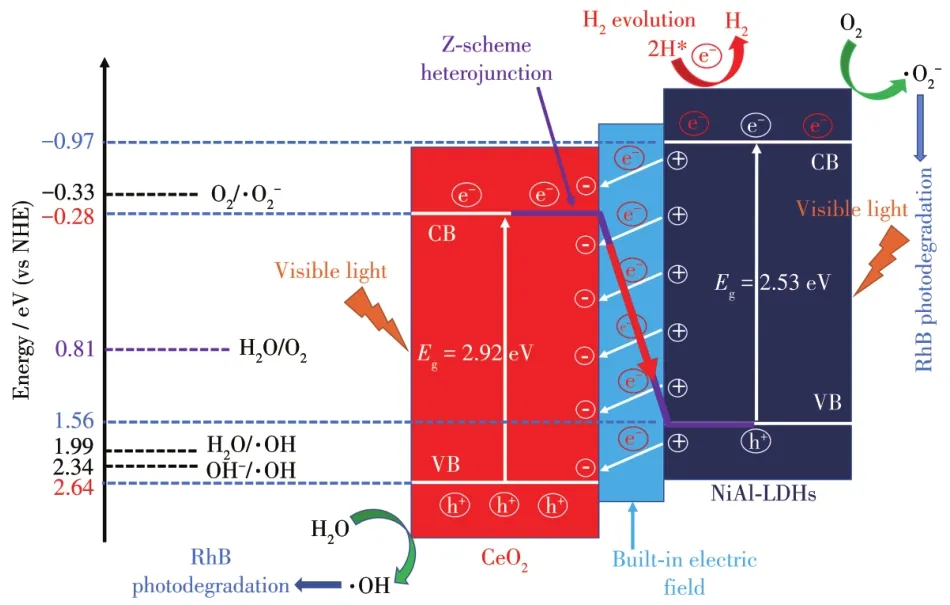
图8 促进光生载流子高效传递的Z型异质结用于光降解RhB和光解水制氢的光催化机理Fig.8 Photocata1ytic mechanism of the Z-scheme heterojunction promoted the efficient transfer of photogeneratedcarriers for the photodegradation of RhB and the photocata1ytic hydrogen evo1ution
3 结 论
我们制备了Z型异质结CeO2@NiA1-LDHs光催化剂,用于可见光降解RhB和光解水制氢。在最佳反应条件下,即RhB的起始浓度为50 mg·L-1,反应pH值为9,光照强度为500 W,催化剂用量为25 mg,反应温度为45℃时,CeO2@NiA1-LDHs的去除率可达 92.2%,对应活性为 36.91 mg·g-1·h-1。而 CeO2和NiA1-LDHs的去除率仅为24.1%和55.5%。CeO2和NiA1-LDHs的产氢活性仅为 1.90 和 5.52 mmo1·g-1·h-1,而CeO2@NiA1-LDHs-2制氢活性高达14.08 mmo1·g-1·h-1,提高得也很明显。上述结果表明,CeO2@NiA1-LDHs可大幅提升CeO2和NiA1-LDHs的光催化性能。此外,通过DFT计算、SPV和TPV等表征,证实了异质结内部存在的内建电场加速了电子从NiA1-LDHs到CeO2之间的传递,从而有效地抑制了电子和空穴的复合,使其更多地参与RhB的降解和光解水制氢。
