键长和电子效应共同调控的β-二酮Dy(Ⅲ)配合物的合成、结构与磁性
2021-01-29董艳萍李光明
董艳萍 李光明
(1黑龙江大学,功能无机材料化学教育部重点实验室,哈尔滨 150080)(2绥化学院食品与制药工程学院,绥化 152061)
单分子磁体可以通过化学设计合成方法获得,其具有可控性好、尺寸完全均一等特点,在高密度信息存储等方面具有独特的应用,一经出现立刻引起化学、物理以及材料学等多学科科学家们的广泛关注[1-7]。单分子磁体以分子作为基本结构单元,且在磁学意义上几乎没有相互作用。在磁阻塞温度(TB)以下呈现的缓慢弛豫行为正是单分子磁体作为存储材料使用的优势之一。稀土离子因其独特的电子结构,如:较多的单电子数、较大的自旋基态、高自旋轨道耦合,所以引入稀土离子来制备高的有效翻转能垒的单分子磁体配合物,成为了科学工作者的研究重点[8-12]。尤其是Dy(Ⅲ),具有大的未猝灭的轨道角动量、Ising型的轴各向异性和|±15/2〉Kramers双重态,因而成为单分子磁体配合物的研究热点[13-17]。
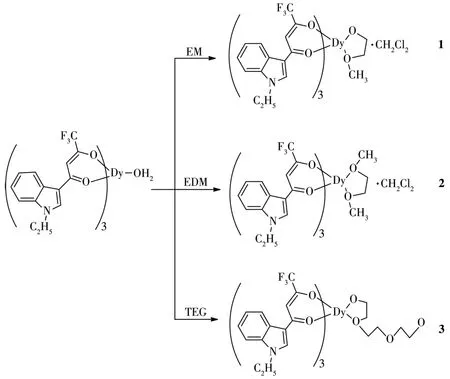
在Dy(Ⅲ)的单分子磁体配合物中,已报道了大量含有不同辅助配体的β-二酮Dy(Ⅲ)配合物,并提出了配合物的配位数、配位场的轴对称性、配位多面体的几何构型等与磁性之间的关系[18-20]。然而对辅助配体的研究也仅仅只集中在含氮辅助配体上,关于辅助配体的电子效应对磁性的影响还尚未见报道。因此人们期望通过改变辅助配体的类型,比如选择含氧辅助配体,进而改变其电子效应,得到磁学性能优良的单分子磁体稀土配合物。我们以N-乙基-3-吲哚三氟甲基β-二酮(EIFD)为主配体,分别选用不同的含氧辅助配体:乙二醇单甲醚(EM)、乙二醇二甲醚(EDM)、二缩三乙二醇(TEG),成功地合成了3个配合物1~3,并确定了它们的单晶结构,研究了它们的磁性。
1 实验部分
1.1 实验仪器与试剂
所用仪器有:Quantum Design SQUID MPMSXL7磁强计、Oxford Xca1ibur Gemini U1tra X射线单晶衍射仪、Raxis-Rapid X射线粉末衍射仪(CuKα,λ=0.154 18 nm,扫描范围为 5°~50°,加速电压为 40 kV,工作电流为40 mA)、Perkin-E1mer 2400元素分析仪、Perkin-E1mer Lambda 35紫外光谱仪、Perkin-E1mer 100红外光谱仪、Perkin-E1mer STA 6000差热/热重热分析仪。
EM、EDM、TEG、二氯甲烷、正己烷均购自天津市科密欧化学试剂有限公司。[Dy(EIFD)3(H2O)]·CH2C12的合成方法见文献[21]。
1.2 配合物的合成
[Dy(EIFD)3(EM)]·CH2C12(1):在试管中将[Dy(EIFD)3(H2O)]·CH2C12(0.111 0 g,0.1 mmo1)溶于CH2C12,再向上述溶液中滴加适量的EM,利用溶剂(正己烷)扩散法,3 d后即可得到无色的配合物晶体。产率:82%。元素分析(C181H160C12Dy4F36N12O32,4 420.12)计算值(%):C,49.18;H,3.62;N,3.80。实测值(%):C,49.20;H,6.63;N,3.82。红外光谱(KBr,cm-1):3 343(m),1 601(s),1 531(s),1 452(m),1 382(s),1 281(s),1 218(s),1 126(s),798(s),751(s),665(s)。
Scheme 1 Synthesis routes of comp1exes 1~3
[Dy(EIFD)3(EDM)]·CH2C12(2):在试管中将[Dy(EIFD)3(H2O)]·CH2C12(0.111 0 g,0.1 mmo1)溶于CH2C12,再向上述溶液中滴加适量的EDM,利用溶剂(正己烷)扩散法,5 d后即可得到无色的配合物晶体。产率:81%。元素分析(C47H43C1DyF9N3O8,1 146.80)计算值(%):C,49.22;H,3.75;N,3.66。实测值(%):C,49.20;H,3.76;N,3.68。红外光谱(KBr,cm-1):3 414(m),1 602(s),1 537(s),1 454(m),1 383(s),1 282(s),1 219(s),1 125(s),798(s),752(s),666(s)。
[Dy(EIFD)3(TEG)](3):在试管中将[Dy(EIFD)3(H2O)]·CH2C12(0.111 0 g,0.1 mmo1)溶于CH2C12,再向上述溶液中滴加适量的TEG,利用溶剂(正己烷)扩散法,3 d后即可得到无色的配合物晶体。产率:83%。元素分析(C96H90Dy2F18N6O20,2 314.75)计算值(%):C,49.81;H,3.89;N,3.63。实测值(%):C,49.80;H,3.88;N,3.65。红外光谱(KBr,cm-1):3 416(m),1 602(s),1 537(s),1 453(m),1 384(s),1 281(s),1 218(s),1 126(s),798(s),751(s),665(s)。
1.3 配合物晶体结构的测定
采用Oxford Xca1ibur Gemini U1tra X射线单晶衍射仪,选取一定尺寸的配合物单晶,辐射源为MoKα(λ=0.071 073 nm),在室温下收集单晶衍射数据。通过差值Fourier合成法进行结构解析,运用SHELXS-2014程序包锁定和修正[22],通过经验吸收校正和Lp因子校正。配合物1和2利用SQUEEZE处理,忽略了溶剂分子。配合物1~3的晶体学数据见表1。
CCDC:1893554,1;1893555,2;1893556,3。
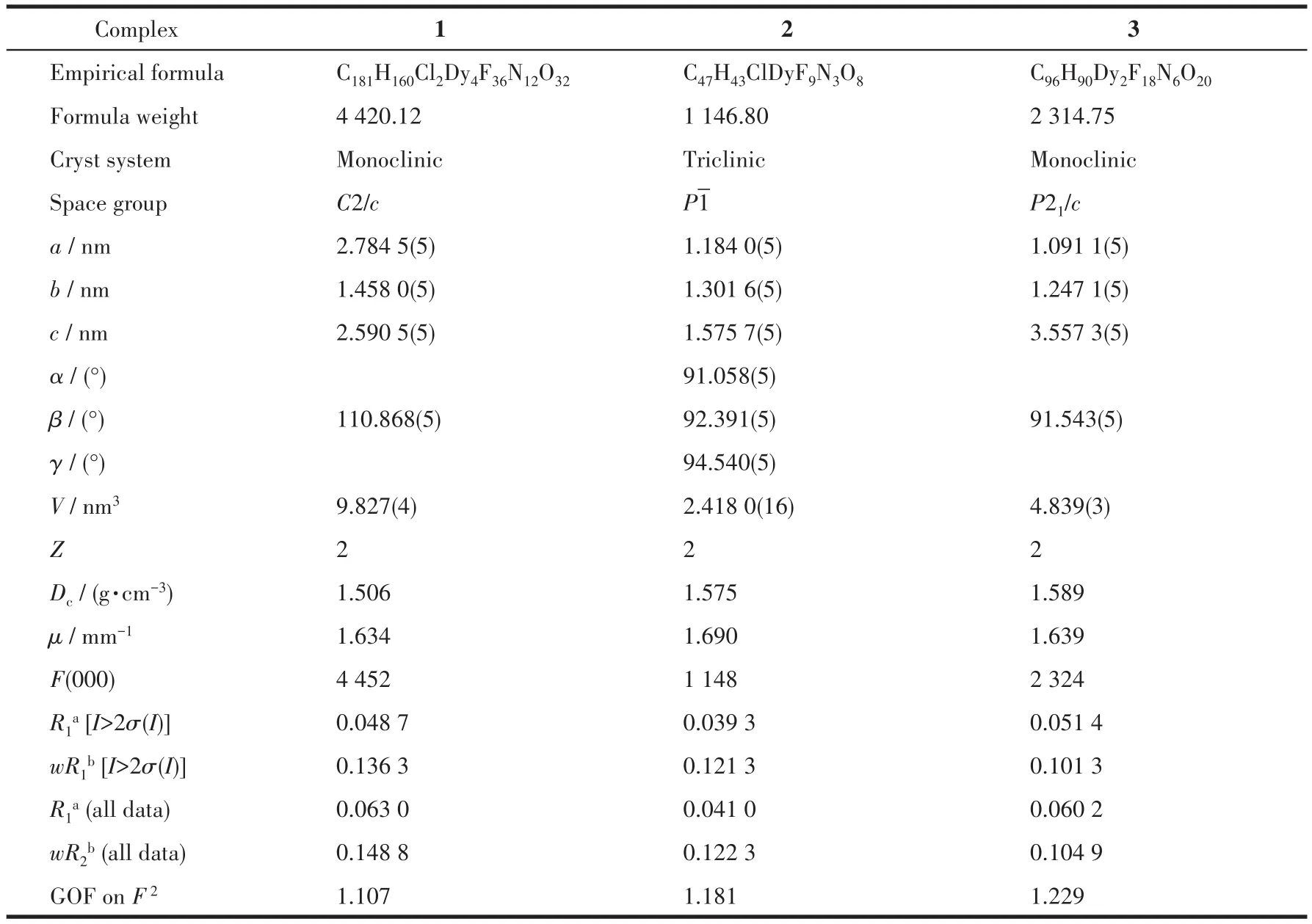
表1 配合物1~3的晶体学精修数据Table 1 Crystal data and structure refinement for complexes 1~3
2 结果与讨论
2.1 配合物的晶体结构
配合物1和3均属单斜晶系,配合物2属三斜晶系,配合物1是C2/c空间群,配合物2是P1空间群,配合物3是P21/c空间群。配合物1~3的分子结构及配位构型如图1所示,均为单核八配位结构,与中心Dy(Ⅲ)配位的氧原子,分别来自3个配体EIFD的6个氧原子和辅助配体 EM(1)、EDM(2)和 TEG(3)的 2个氧原子,Dy—O平均键长为分别为0.237 5、0.237 1和0.237 9 nm。通过SHAPE 2.1软件计算规则得出的数值(表2)越小,配合物越接近于理想多面体的配位构型[23],据此判断配合物1~3的配位构型分别为双帽三棱柱、正十二面体和双帽三棱柱,分别具有C2v、D2d和C2v对称性。
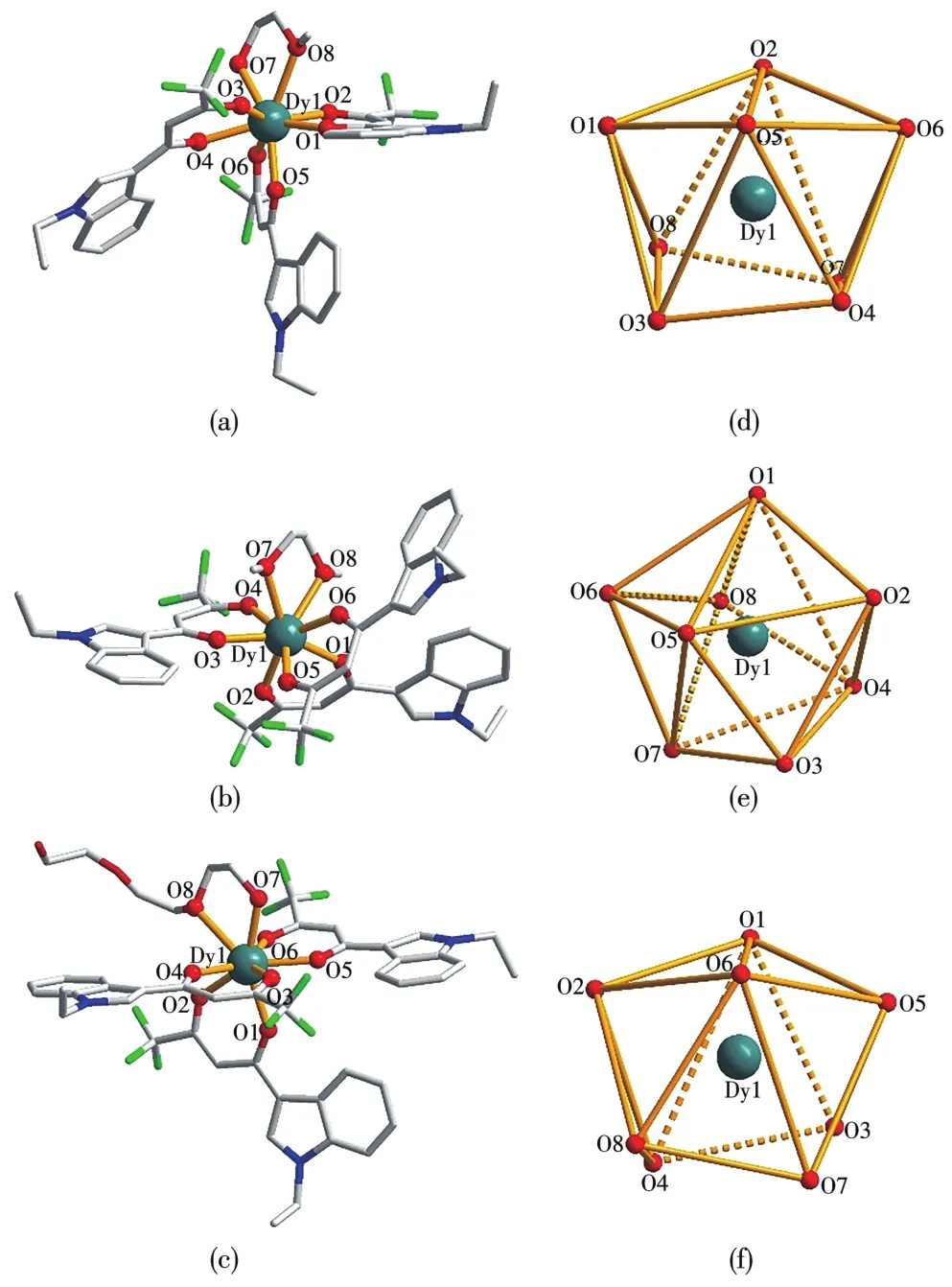
图1 配合物1~3的分子结构图(a~c)和配位构型图(d~f)Fig.1 Crysta1 structure(a~c)and 1oca1 coordination geometry(d~f)for comp1exes 1~3

表2 配合物1~3的Shape分析Table 2 Shape analysis of complexes 1~3
2.2 配合物的X射线粉末衍射分析
配合物1~3的X射线粉末衍射(XRPD)分析结果如图2所示,测试结果显示:主要峰位与单晶CIF文件模拟的峰位吻合度较好,与测定的单晶数据结构相同,进一步表明了宏量制备的配合物样品为纯相,且具有均相性。

图2 配合物1~3的XRPD图Fig.2 XRPD patterns for comp1exes 1~3
2.3 配合物的差热/热失重分析
在30~800℃温度范围内,氮气保护下,配合物1~3的差热/热失重(DSC/TG)分析如图3所示。其中配合物1和2的第一步失重在90~100℃之间,归因于溶剂分子CH2C12的失去,计算值分别为3.84%(1)和 7.41%(2),实测值分别为 3.07%(1)和 7.58%(2),计算值和理论值相符,而配合物3在此温度间无失重现象,证明配合物3分子结构中没有溶剂分子。随着温度的升高达到300℃左右时,配合物1~3均开始发生骨架分解。温度继续升高,持续发生失重。当温度达到550℃左右时,配合物骨架完全坍塌,最终分解成稀土氧化物。

图3 配合物1~3的DSC/TG曲线Fig.3 DSC/TG curves for comp1exes 1~3
2.4 配合物的分子磁性研究
在1 000 Oe的外加磁场下,测试了配合物1~3在1.8~300 K的变温磁化率。如图4所示,配合物1~3的χmT值略低于Dy(Ⅲ)离子的理论值14.17 cm3·K·mo1-1,分别为 14.12、14.11 和 14.12 cm3·K·mo1-1,并随着温度的降低而逐渐下降。由于不断减少的斯塔克效应和分子间偶极-偶极相互作用[24-25],温度降至80 K左右时,χmT值的下降开始变快。1.8 K时,χmT值达到最小,分别为9.63、11.13和11.28 cm3·K·mo1-1,归因于塞曼分裂晶体场能级的减少[26]。在不同温度(1.8、3和5 K)下(图4中内插图),M-H曲线不重合,说明了配合物中Dy(Ⅲ)的磁各向异性和/或低激发态[27]。同时对配合物1~3进行磁滞回线测试,如图5所示,配合物1和3有明显的蝴蝶状磁滞回线,而配合物2无明显的现象。
在零场下测试了配合物1~3在频率1~1 000 Hz的交流磁化率温度依赖曲线,如图6所示,由于量子隧穿效应的影响,只有配合物1和3的实部和虚部都出现了频率依赖现象,但是没有出现明显的频率依赖最大值;配合物2的虚部出现了较弱的频率依赖现象,同样也没有出现明显的频率依赖最大值。
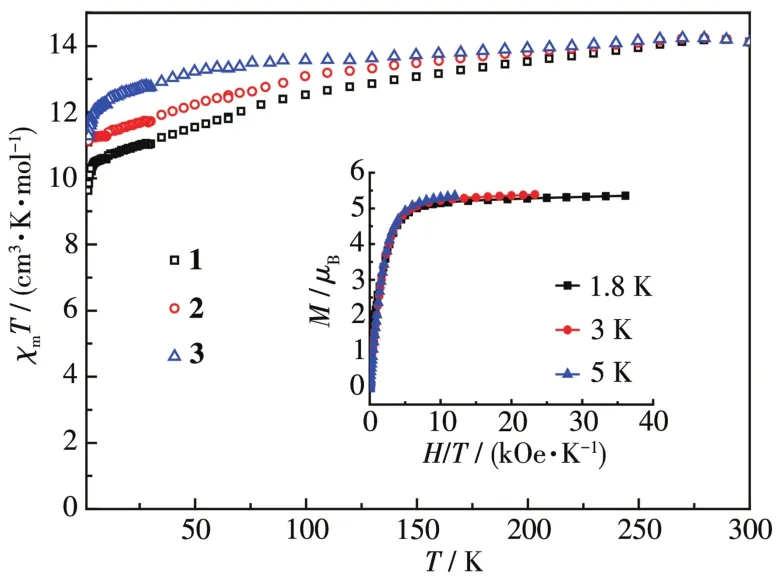
图4 在场强1 000 Oe下配合物1~3的M-T曲线Fig.4 P1ots of M-T for comp1exes 1~3 under 1 000 Oe fie1d

图5 配合物1和3的磁滞回线Fig.5 Hysteresis 1oop for comp1exes 1 and 3

图6 配合物1~3在零场下的交流磁化率温度依赖曲线Fig.6 Temperature dependence of ac susceptibi1ity for comp1exes 1~3 under 0 Oe fie1d
为了降低量子隧穿效应的影响,外加磁场选为1 500 Oe,可观察到配合物1~3的量子隧穿效应明显减少了,实部和虚部都出现了频率依赖最大值,具有慢弛豫现象,如图7所示。配合物3还出现了明显的双弛豫现象。利用Arrhenius公式τ=τ0exp[Ueff/(kBT)]进行线性拟合,得到配合物1和2的有效翻转能垒Ueff和指前因子τ0分别为95.1 K和1.3×10-6(1)、40.5 K和4.9×10-7(2);得到配合物3的有效翻转能垒Ueff为 53.8 和 13.4 K,指前因子τ0为 7.6×10-7和 5.9×10-6,如图8所示。
在外加磁场1 500 Oe、温度2~18 K下,测试了配合物1~3的交流磁化率频率依赖曲线,如图9所示。配合物的单分子磁体行为也可以通过Co1e-Co1e曲线证实,如图10所示。曲线通过德拜公式拟合得到配合物1和2的参数α值小于0.25,配合物3的α1值在0.61~0.23之间,α2在0.80~0.16之间。弛豫中的高温部分应该属于Orbach过程,低温部分应该包含有Raman过程,配合物1和2具有单一弛豫模式,配合物3存在双弛豫模式,与文献报道的配合物类似[28-30]。
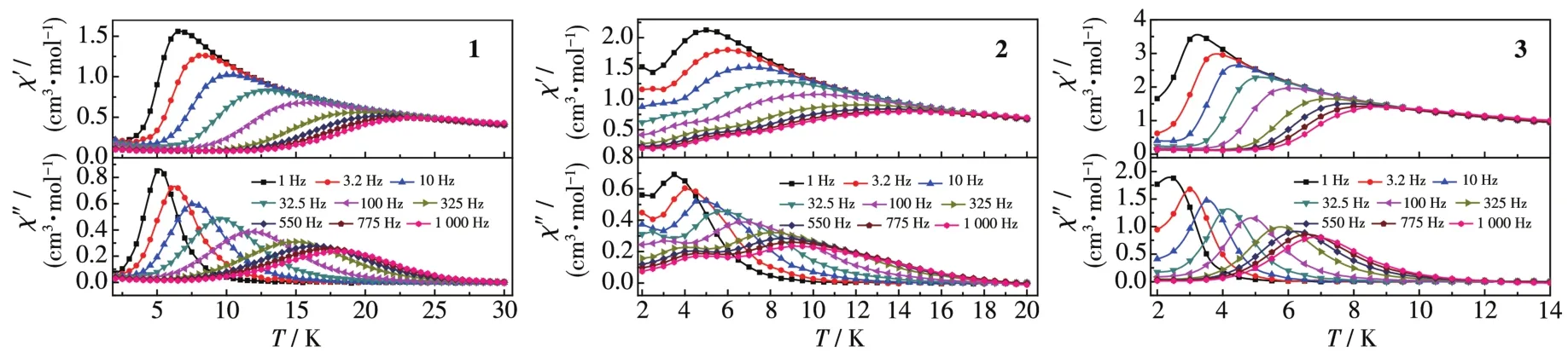
图7 配合物1~3在场强1 500 Oe下的交流磁化率温度依赖曲线Fig.7 Temperature dependence of ac susceptibi1ity for comp1exes 1~3 under 1 500 Oe fie1d
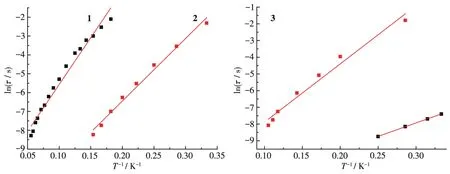
图8 配合物1~3的1n τ-T-1曲线Fig.8 P1ots of 1n τ-T-1for comp1exes 1~3

图9 配合物1~3在场强1 500 Oe下的交流磁化率频率依赖曲线Fig.9 Frequency dependence of ac susceptibi1ity for comp1exes 1~3 under 1 500 Oe fie1d

图10 配合物1~3在场强1 500 Oe下的Co1e-Co1e曲线Fig.10 Co1e-Co1e curves for comp1exes 1~3 under 1 500 Oe fie1d
值得注意的是,配合物1中Dy—O键长明显小于配合物2中Dy—O键长,这将导致不同静电势的分布,有利于Dy(Ⅲ)的|±15/2〉Kramers双重态扁圆形状的稳定,从而产生较强的磁各向异性[31]。同时,配合物1辅助配体EM的吸电子效应强于配合物2辅助配体EDM,同样也会使Dy—O的键长受到不同程度的压缩,键长越短,说明压缩得越明显,越有利于增强磁各向异性[32]。因此,配合物1在1 500 Oe下的有效翻转能垒要高于配合物2,进一步证明了β-二酮Dy(Ⅲ)配合物的磁性不仅与文献报道的配位数、配位场的轴对称性、配位多面体的几何构型等因素有关,还与其他参数如Dy—O键长、辅助配体的电子效应等有重要的关系。
与已报道的含氮辅助配体的β-二酮Dy(Ⅲ)配合物对比,配合物1~3的有效翻转能垒高于[Dy(d-tfc)3(phen)]·2H2O、Dy(l-tfc)3(phen)、[Dy(hfac)3(L)]·C6H14等类似配合物[18-21,33-38]。主要归因于氧原子的电负性强于氮原子,从而导致配合物1~3配体场在对称轴方向上的电子密度有所增强[39],因此,配合物1~3具有较高的有效翻转能垒。
3 结论
合成了3个配体和含氧辅助配体共同参与配位的单核β-二酮Dy(Ⅲ)配合物1~3。磁性研究结果显示,配合物1~3属场诱导的单分子磁体,具有较高的有效翻转能垒。配合物Dy—O的键长和含氧辅助配体对配合物的磁性有重要的调节作用,其中Dy—O的键长越短、含氧辅助配体的吸电子效应越强,配合物越能展现出优良的磁学性能。
