内质网应激及其在糖尿病肾病中的作用机制
2021-01-27金童陈铖
金童,陈铖
武汉大学人民医院肾内科, 武汉 430060
据国际糖尿病联盟最新报告统计,2019年全球约4.63亿成人(20~79岁)患糖尿病,约有420万人(20~79岁)死于糖尿病或其并发症。预计到2030年和2045年糖尿病患者将分别达到5.784亿和7.002亿[1]。糖尿病肾病(DN)是糖尿病最常见的微血管并发症,在过去的十年中DN的发病率不断上升,是全球终末期肾脏疾病(ESRD)的主要原因之一[2]。因此,研究DN的发病机制和相应的治疗措施十分重要。当前已明确糖尿病肾病的发病机制与糖代谢异常、脂代谢紊乱、免疫炎症、氧化应激、内质网应激、凋亡自噬等因素相关[3]。其中内质网应激(ERS)在DN的发生和进展中起着关键的作用,具有很重要的研究前景。ERS通过触发细胞的3条经典的未折叠蛋白反应(unfolded protein response, UPR)信号通路来促进ER处理应激的能力,本质上是一种机体对有害刺激的自身应答反应,然而过强或持续的ERS超越了细胞的耐受能力,将会导致细胞凋亡。本文就DN中ERS激活UPR的调控机制以及二者关系的研究进展进行综述,以期为DN的治疗研究提供参考。
1 内质网应激和未折叠蛋白反应
1.1 内质网稳态及维持
内质网是多功能的膜性细胞器,负责真核细胞中至少三分之一的蛋白质合成、折叠、组装和运输,通过监测所有进入细胞器的蛋白质的生物合成、折叠、组装、运输和降解的过程来调节蛋白质稳态,其稳态的平衡对于细胞的正常生理功能极其重要[3-4];同时内质网也是细胞内钙离子稳态调节及多种脂质(类固醇和胆固醇)合成的重要场所。内质网中一些分子伴侣和酶对于正确的蛋白折叠和内质网正常生物合成至关重要[5]。
内质网稳态参与多种生理和病理过程,当各种刺激因素如营养缺乏、缺血缺氧、感染、氧化应激、内质网钙含量异常、脂质超载等导致内质网稳态失衡时,就会发生内质网应激反应,可激活未折叠蛋白反应、内质网超负荷反应和固醇调节级联反应等信号通路[6-8]。其中 UPR是目前研究最多的通路,可通过调节基因表达程序来提升内质网的应激处理能力,包括以下几个方面:①抑制蛋白质合成以预防细胞被自身合成的错误蛋白质所侵害;②分泌特定的蛋白酶降解错误折叠的蛋白质;③诱导分子伴侣和蛋白质加工酶基因的表达来增强蛋白质的折叠能力;④增加参与脂质代谢基因的表达来促进ER膜扩张,从而增强ER的功能[7-9]。简单来说,这个反应就是细胞对于蛋白质的一个质量控制系统,用来修复/销毁被错误折叠的蛋白质,最终目的是减轻内质网负担与重建内质网稳态。研究表明适宜的刺激可激活UPR启动细胞保护机制以维持内质网稳态,在无法补救的ERS情况下,UPR会转变为另一个信号平台,称为末端UPR,过强或长时间刺激可引发过度UPR激活细胞损伤机制致细胞凋亡,进而导致疾病的发生和进展[10]。
1.2 UPR与内质网应激
哺乳动物中,UPR由分子伴侣葡萄糖调节蛋白78(GRP78)/免疫球蛋白结合蛋白(BIP)及3种内质网跨膜蛋白所介导,后者分别是:肌醇需求酶1 (inositol-requiring enzyme, IRE1)、双链RNA依赖的蛋白激酶样内质网激酶(double stranded RNA-dependent protein kinase-like endoplasmic reticulum kinase,PERK)和活化转录因子6 (activating transcription factor 6, ATF6)[6]。正常情况下,GRP78与上述3种蛋白的腔结构域结合,处于失活状态,当发生ERS时,3种酶与BIP解离后被活化,分别激活下游生存信号通路。然而ERS是一把双刃剑,当细胞处于过度或持续的ERS下,促凋亡转录因子如CHOP、c-JNK、Capase-12被激活,细胞的凋亡程序启动[11-12]。
1.2.1IRE-1信号通路 IRE-1是位于内质网膜上的Ⅰ型跨膜蛋白, 具有核酸内切酶活性的胞质段结构域,介导最保守的一条UPR信号通路[13]。ERS发生时,IRE-1与GRP78解离,与错误折叠蛋白结合,其N端发生二聚化并触发了自身的磷酸化,C端的核酸内切酶活性被变构激活,活化后的IRE-1与XBP1(X box binding protein-1) 前体mRNA结合,剪切26个碱基内含子组成的片段产生新的mRNA,翻译成活跃且稳定的转录因子XBP1s(X box binding protein 1 splicing)[7,14]。XBP1s和ERS反应元件结合诱导激活内质网相关性降解因子(endoplasmic reticulum-associated degradation,ERAD)和内质网分子伴侣蛋白的表达,从而促进细胞的适应性生存[15]。
1.2.2PEPK信号通路 PERK与IRE-1也是位于内质网膜上的Ⅰ型跨膜蛋白,含有腔内应激感应结构域和胞质内丝氨酸/苏氨酸蛋白激酶功能域[16]。发生ERS时,游离的PERK发生激酶结构域的同源二聚化和自身磷酸化而激活,通过使真核翻译起始因子2α(eIF2)51位的丝氨酸磷酸化, 减慢了整体蛋白质的翻译速度,使细胞有更多的时间折叠积压在ER内腔中的蛋白质[17],这种翻译抑制作用对于维持胰腺β细胞存活和代谢稳态至关重要[18]。此外, 磷酸化的eIF2α能够激活活化转录因子4(ATF4)的翻译, 从而上调许多UPR目标基因的翻译水平以增强内质网的蛋白折叠能力、抗氧化反应和自噬能力[19]。
1.2.3ATF6通路 ATF6是一种Ⅱ型ER跨膜蛋白,包括腔内C末端、跨膜区、胞质N末端3个结构域。在胞质中包含bZIP转录激活域,在ER内腔中包含压力感应结构域[13]。发生ERS时,ATF6与GRP78解离后被转运到高尔基体,由高尔基体蛋白酶S1P及S2P对其跨膜片段进行切割,产生游离的50 kD大小的N端片段,转移到细胞核后结合内质网应激反应元件(ERSE)上调GRP78、GRP94和Calreticulin等分子伴侣和XBP1基因的表达[20-21]。此外,ATF6可通过与XBP1s形成异源二聚体上调内质网相关的蛋白质降解因子、分子伴侣、糖基化酶,细胞内转运机制和蛋白质二硫键异构酶来增强ER蛋白的折叠能力的表达,缓解ERS的压力[22-23]。
1.3 UPR介导的凋亡途径
细胞凋亡有3条途径:线粒体/细胞色素c介导的凋亡途径、死亡受体介导的凋亡途径和ERS诱导性凋亡途径[19]。近年来,诱导性途径ERS逐渐受到关注,主要有3条信号通路参与(图1):①CHOP/GADD153信号通路:持续活化的PERK通过促进ATF4 mRNA的转录表达,上调CHOP基因,并通过ROS产生和ATP消耗促进细胞凋亡[24]。过表达的CHOP可以下调抗凋亡基因Bcl-2的表达,加速细胞凋亡。同时,CHOP可激活GADD34,促进磷酸化真核翻译起始因子2α亚基(p-eIF2α)去磷酸化,形成PERK通路的负反馈调节,使蛋白合成暂停得以恢复[25]。此外,有研究指出,CHOP可下调抗凋亡蛋白BCL2、BCL-XL和MCL-1的表达,并上调BIM的表达,从而促进BAK和BAX的表达。BAX-BAK发生寡聚化后,寡聚体通过线粒体透化作用导致凋亡因子如细胞色素c(Cyt c)和凋亡诱导因子(AIF)释放,最终导致细胞死亡。②JNK信号通路:活化的IRE-1α与TRAF2结合后激活ASK1,三者形成复合物后进一步激活JNK,上调CHOP、Bcl-2 家族中的促凋亡因子PUMA、BID和BIM,同时下调抗凋亡基因Bcl-2、Bcl-XL,引发线粒体途径介导的细胞凋亡。同时IRE1α-TRAF2也能通过钙调蛋白分解酶活化Caspase-12,激活Caspase介导的凋亡通路[26-27]。③Caspase-12信号通路:Caspase激活是一个级联放大反应,ERS时,内质网释放Ca2+进入细胞质中,刺激周围钙蛋白酶(Calpain)的活化并转移到内质网外膜,同时引发Caspase-7转位后活化,两者均可将Procaspase-12水解为活化的Caspase-12进入胞浆发挥作用,并可通过细胞色素c非依赖途径激活Caspase-9、Caspase-3等,引发细胞凋亡[28]。
2 ERS参与DN发生发展的机制研究
DN的临床表现主要是蛋白尿、水肿、肾功能减退,形态学表现为肾小球细胞外基质堆积、系膜扩张、基底膜增厚、足突融合、肾小球硬化及肾小管间质纤维化。ERS与DN的发生发展密切相关,研究证明DN的很多属性,如高血糖症、蛋白尿、晚期糖基化终产物和游离脂肪酸的增加,都可
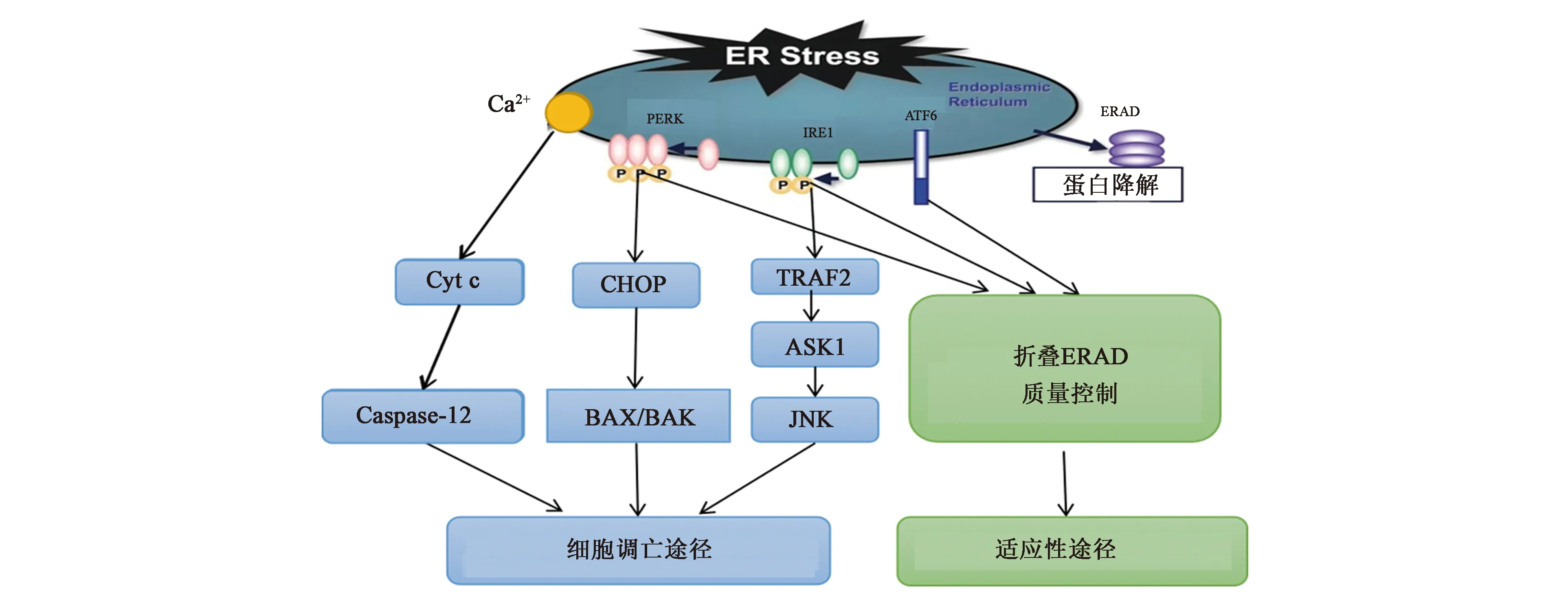
图1 UPR介导的细胞适应性途径和凋亡途径Fig.1 UPR mediated adaptive and apoptotic pathways of cells
以触发肾细胞中的未折叠蛋白反应。在1型糖尿病的链脲佐菌素模型中,肾小球和肾小管细胞中BIP、p-PERK、p-JNK、CHOP/GADD153和Caspase-12的水平升高有关。在2型糖尿病肾病的db/db小鼠模型中,内质网应激通过XBP1s触发了炎症基因的表达。下面介绍ERS在不同肾细胞类型中的相关研究。
2.1 参与肾小球足细胞损伤
足细胞是终末分化的肾小球上皮细胞,再生能力有限。据报道,蛋白尿是DN肾小球功能障碍的原因,ERS是白蛋白引起足细胞损伤的重要机制之一。Goncalves等[29]研究表明GRP78/IRE1-α/PKC-δ/Caspase-12信号通路参与了ERS和诱导白蛋白超负荷依赖性足细胞损伤和凋亡。CHOP是导致凋亡激活的关键ERS转录因子,Fan等[30]研究发现,网状蛋白1A(RTN1A)诱导PERK磷酸化从而诱导CHOP表达,CHOP的敲低显著抑制了RTN1A的表达,RTN1A和CHOP之间的正反馈回路导致足细胞ERS增强,RTN1A可能是足细胞ERS的关键调节因子。长基因间非编码RNA(LINC01619)在肾脏中主要分布在足细胞细胞质,通过充当miR-27a的“海绵”发挥生物学功能。高糖培养的足细胞中,LINC01619表达下调,对miR-27a的吸附减少,负向调控叉头盒蛋白O1(FOXO1)激活ERS介导的凋亡途径并导致足细胞损伤,因此LINC01619可作为竞争性内源RNA调节DN中miR-27a/FOXO1介导的内质网应激和足细胞损伤[31]。
2.2 参与肾小球系膜细胞(GMCs)损伤
肾小球系膜细胞是DN进展的重要标志。在高糖刺激肾小球系膜细胞内,miR-148b、GRP78、CHOP表达均显著上调,靶向抑制AMPKα1的表达,从而诱导ERS介导的凋亡途径,使肾小球系膜细胞产生过多细胞外基质蛋白[32]。脂毒性是DN恶化的主要原因之一,可通过PERK和ATF6信号通路诱导肾小球系膜细胞凋亡。Park等[33]研究表明,蛋白质精氨酸甲基转移酶1(PRMT1)介导模拟细胞脂毒性的棕榈酸酯诱导的ERS凋亡途径致肾小球系膜细胞凋亡,降低PRMT1表达或降低其酶活性的策略可用于预防糖尿病性肾病的恶化。此外,研究表明脂肪酸结合蛋白4(FABP4)主要在人肾活检的肾小球系膜细胞中表达,在DN中,FABP4的表达上调伴随着GRP78和Caspase-12的上调,而抗凋亡蛋白Bcl-2的表达下调,导致系膜细胞凋亡[34]。
2.3 参与肾小球内皮细胞(GECs)损伤
高糖诱导的ERS与糖尿病患者内皮细胞功能障碍的各个方面密切相关。GECs损伤是糖尿病的主要事件,导致多种大血管和微血管并发症。血管紧张素Ⅱ(AngⅡ)可以拮抗Ang 1与Tie2受体结合,血管生成素1(Angpt1)显著降低了AngⅡ诱导的ERS反应蛋白GRP78、GRP94、p-PERK和CHOP的表达[35]。此外,Angpt1通过GECs中的Tie2受体/ERK1/2-p38 MAPK途径减轻了ERS诱导的细胞损伤和凋亡[36]。
2.4 参与肾小管上皮细胞(TECs) 损伤
肾小管上皮细胞暴露于高浓度人血清白蛋白后可呈时间、剂量依赖性上调GRP78的表达,激活PERK-CHOP凋亡通路,细胞凋亡也呈进行性增加。蛋白尿会促进糖尿病肾病的发展,并诱发ERS和肾上皮小管细胞上皮-间质转化(EMT)[37]。在DN患者肾活检的肾小管间质中UPR相关基因显著上调,DN的肾损伤伴随着Caspase-12活化和肾小管细胞凋亡[38]。在高葡萄糖处理的肾小管上皮细胞中,ATF6的抑制而非PERK的抑制会阻止PRMT1诱导的EMT。此外,Pang等[39]发现尿激肽原Ⅱ(UrotensinⅡ)可能通过触发ERS途径,上调GRP78、CHOP的表达诱导肾小管上皮细胞EMT并增加细胞外基质的生成。
3 通过调控ERS治疗DN的研究进展
近年来,研究者们在ERS途径的调控方面展开了大量的研究,针对ERS的靶点进行干预治疗DN或成为具有潜力的新方法(图2)。被称为“人工合成分子伴侣”的小分子化合物如4-苯基丁酸(4-PBA)和牛磺酸熊去氧胆酸(TUDCA)是经典的ERS抑制剂,可通过阻断ERS介导的凋亡途径PERK-eIF2α-CHOP通路来预防AGEs诱导的足细胞凋亡。有研究证实在用TUDCA处理的db/db-Unx小鼠中,RTN1A以及GRP78、p-PERK和CHOP的表达在蛋白质和mRNA水平上均受到抑制,这表明TUDCA在DN中通过抑制足细胞中的RTN1A和ERS而发挥保护作用。Cao等[40]研究发现TUDCA和4-PBA均可下调ERS相关蛋白BiP、p-PERK、p-IRE1α、ATF-6、XBP-1、Caspase-12和Caspase-3的表达,从而在体外抑制UPR,恢复葡萄糖耐量和改善胰岛素敏感性,逆转肾小球系膜扩张,减少蛋白尿,调节自噬体数量来预防DN的发展。使用TUDCA进行治疗,还可以减轻肾小管间质纤维化,改善糖尿病肾病,包括尿白蛋白和肌酐比值和尿白蛋白排泄率,并减少TECs的凋亡[41]。Fang等[42]研究表明在肾小球系膜细胞中,FABP4抑制剂BMS309403通过FABP4减弱了ERS标志物GRP78、CHOP、Caspase-12的诱导从而减轻了细胞凋亡。依达拉奉是一种有效的自由基抑制剂,在临床上作为脑保护剂,可通过p-eIF2α和CHOP抑制作用来防止脂质过氧化和ERS引起的缺氧,在防治DN方面的作用值得进一步研究[43]。最近,Chu等[44]发现阿司匹林可能通过部分阻断PERK通路,从而抑制高脂血症诱导的足细胞ERS。此外,二甲双胍可以通过激活单磷酸腺苷激活的蛋白激酶(AMPK)途径,减弱高糖诱导的肾小管上皮细胞内质网应激和肾纤维化[45]。我国的一些传统中医药也可通过干预ERS靶点来预防肾脏损害,例如,番红花和槲皮素通过抑制ROS介导的ERS途径来预防细胞凋亡[44];黄芪甲苷 IV(AS-IV)可能通过下调p-PERK、ATF4和CHOP的表达来抑制内质网应激诱导的足细胞凋亡[45];大黄素通过抑制PERK-eIF2α信号传导通路来减轻足细胞的凋亡[44]。要确定这些药物在多大程度上可以缓解肾脏ERS减少肾脏损伤,仍需要进一步的评估研究。

图2 靶向内质网应激治疗糖尿病肾病的分子机制Fig.2 Molecular mechanism of targeting endoplasmic reticulum in emergency treatment of diabetic nephropathy
4 展望
ERS/UPR对于细胞生命活动的调节是一把双刃剑,对于细胞来说既是一种反应性保护机制,又可以启动凋亡程序,所以调控两者之间的平衡并充分发挥保护作用仍是需要不断研究的方向。同时,由于很多作用于ERS的药物缺乏特异性、安全性和有效性,因此需要加强开发和评估针对ERS途径的特定分子的无毒性药。DN中的多种因素诸如氧化应激、自噬等可导致ERS激活,最新有文献报道铁死亡、炎症小体等机制也可能与ERS偶联,深入探究ERS与其他作用机制特定效应分子、信号通路的相关性,将为以后的临床治疗和预防提供新思路。
