内质网自噬
——疾病防治的新靶标
2015-06-09李赫宁李兰芳陈临溪
李赫宁,李兰芳,陈临溪
(南华大学药物药理研究所,湖南 衡阳 421001)
内质网自噬
——疾病防治的新靶标
李赫宁,李兰芳,陈临溪
(南华大学药物药理研究所,湖南 衡阳 421001)
内质网自噬是指内质网功能发生改变时,细胞激活选择性自噬以清除细胞内受损的内质网或内质网片段的过程。其主要的功能是改善细胞内环境,起细胞保护作用。内质网自噬作为选择性自噬研究的新领域,与细胞中其他类型的选择性自噬和细胞凋亡之间关系密切,在疾病发生发展中有重要作用。该文综述了内质网自噬的形成过程,细胞内调控方式,与其他选择性自噬间的相互作用,并阐述了内质网自噬参与糖尿病、神经退行性疾病和肾脏疾病等多种疾病的发生发展,逐步成为疾病防治的新靶标。
内质网自噬;自噬;未折叠蛋白反应;内质网应激;选择性自噬;药物靶标
自噬(autophagy),也被称为“自我吞噬”,是细胞降解和回收利用细胞内生物大分子与细胞器的过程。自噬主要分为3类:大自噬(macroautophagy)、微自噬(microautophagy)和分子伴侣介导的自噬(chaperone-mediated autophagy)。自噬普遍存在真核细胞中,不仅是细胞应对不良环境自发产生的一种保护机制,也参与多种疾病的病理过程[1]。
以往研究认为,自噬是非特异性的降解细胞器或其他细胞组分的过程。最新研究发现,部分自噬过程也具有选择性,当细胞内细胞器发生损伤或功能缺失时,细胞器或细胞器的一部分将会被自噬体吞噬,再与溶酶体融合发生降解,该过程被称为“选择性自噬 (selective autophagy)”。Bernales等[2]发现内质网自噬(reticulophagy)能有效清除细胞内受损的内质网,消除未折叠蛋白反应(unfolded protein response,UPR)和降解蛋白聚集体,有助于内质网中蛋白的正确折叠,维持细胞内环境平衡。研究提示内质网自噬参与人类多种疾病,如糖尿病、神经退行性疾病和肾脏疾病等的发生发展,可能成为疾病防治的新靶标。
1 内质网自噬的定义
Kraft等[3]发现在饥饿状态下,真核细胞中成熟的核糖体会通过选择性自噬快速降解,该过程被称为“核糖体自噬(ribophagy)”。Lemasters等[4]进一步提出“线粒体自噬(mitophagy)”的概念:线粒体在氧化应激、营养缺乏和细胞衰老等刺激作用后发生去极化,进而被特异性自噬膜包裹并与溶酶体融合,该过程在维持细胞稳态的过程中发挥重要作用。除此之外还有“分泌自噬(crinophagy)”[5]、“过氧化物酶体自噬(pexophagy)”[6]、“脂自噬(lipophagy)等”[7]。
Bernales等[2]用电子显微镜观察酵母中内质网结构时发现,UPR引起内质网重构,也会诱导出现双层膜结构大囊泡。该囊泡被证实是只含有内质网的自噬体,因此该囊泡形成和降解的过程被称为“内质网自噬”。所谓内质网自噬是指当细胞内外环境改变时,内质网中未折叠蛋白或错误折叠蛋白增多,引发内质网应激,使内质网功能发生改变,破坏细胞内稳态,继而激活细胞选择性自噬以清除细胞内受损的内质网或内质网片段[2]。
2 内质网自噬的形成
自噬体是如何将内质网选择性吞噬并降解的?主要通过两种途径:(1)含未折叠蛋白或蛋白聚集体的内质网的片段从内质网上脱离,然后被自噬体识别并吞噬(Fig 1A)。(2)自噬体直接在内质网形成或激活,直接包裹要被降解的内质网(Fig 1B)。与前者相比,后者发生过程更具有选择性,该过程不需要特定的细胞组件去引导内容物被自噬体吞噬,而是在内容物膜上募集自噬相关蛋白,作为自噬体膜的源头引发自噬。在自噬体形成起始,ATG1/ULK(autophagy associated gene 1,ATG1,在酵母中命名为ATG1,其哺乳动物同源蛋白命名为ULK)复合物的活性对协调ATG蛋白有重要作用[8]。在正常情况下,哺乳动物雷帕霉素靶蛋白复合物1(mammalian target of rapamycin complex 1,mTORC1)与ATG1/ULK复合物结合,从而抑制自噬。在自噬诱导条件下,mTORC1与ATG1/ULK复合物分离,促使自噬的成核和延伸[9]。
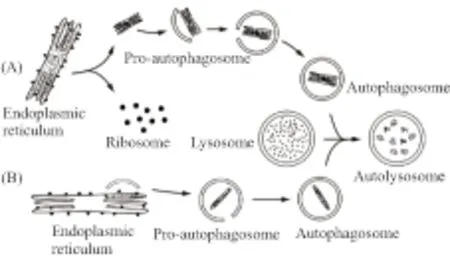
Fig 1 Schematic diagram of reticulophagy processes
关于自噬体膜起源有很大的争议,很多学者认为膜起源于内质网和线粒体[10]。Hamasaki等[11]发现在哺乳动物细胞中自噬体形成于内质网和线粒体的连接位点,其结果显示饥饿处理后,自噬体的标志物ATG14重定位在内质网与线粒体的连接位点,自噬体形成前的标志物ATG5也定位在该位点,内质网蛋白Syntaxin17偶联ATG14也募集其于该位点。Bernales等[2]发现包含内质网的自噬体膜上常镶嵌着核糖体,因此他们认为自噬体膜来自内质网,也就是说内质网自噬是内质网自我吞噬的过程。Lipatova等[12]认为,三磷酸鸟苷(GTP)结合蛋白基因(Ypt1)负责调节内质网向高尔基体转运,也可能调控内质网转化成自噬体,控制内质网自噬体的发生过程,即内质网自噬体膜是由内质网膜转运而形成的。Axe等[13]报道了DFCP1(double FYVE domain-containing protein 1)可能是自噬体膜的起源位点,DFCP1位于内质网膜和高尔基体膜上,也可作为内质网自噬的标志物[14]。因此内质网自噬过程中,ATG组件可能催化分离受损内质网片段或安排一个已经存在的内质网膜构成自噬体的新膜。
目前主要通过荧光显微镜和电子显微镜观察自噬体中是否含有内质网膜来判断是否属于内质网自噬。使用还原剂二硫苏糖醇(DL-dithiothreitol,DTT)处理酵母,通过抑制蛋白二硫键的形成,导致蛋白的错误折叠,可以观察到自噬体中包裹着大量内质网膜结构。当酵母中葡萄糖缺乏引起内质网应激时,糖蛋白N-糖基化受阻,可以观察到自噬体中会存在单一的内质网片段[15]。电子显微镜超微结构分析证实内质网存在于自噬体中。哺乳动物体内α1-抗胰蛋白酶Z突变体的研究中,该突变体在内质网聚集,而胞质中的突变体聚合物与绿色萤光蛋白-微管相关蛋白1轻链3(microtubule-associated protein light chain 3,LC3)和内质网的KDEL蛋白共定位[16]。荧光显微镜也可以检测到酵母发生内质网自噬时ATG8与内质网标志蛋白Sec61共定位。通过对内质网钙网蛋白(calreticulin)免疫标记,也能检测自噬体是否包含内质网片段,判断细胞是否发生内质网自噬[17]。
3 内质网自噬的激活
作为细胞重要的细胞器之一,内质网稳态改变会引发一系列细胞反应,包括UPR、内质网应激以及内质网自噬等。内质网负责分泌蛋白和膜蛋白的折叠、加工和修饰。当受到外界刺激因素的作用导致内质网功能失衡,使内质网中未折叠蛋白和错误折叠蛋白大量累积,就会引发内质网损伤,形成内质网应激(endoplasmic reticulum stress)。内质网应激导致细胞激活自我保护机制,如增强蛋白折叠能力、减少蛋白的翻译和加速蛋白质的降解等[18-19],该过程被称为未折叠蛋白反应,即当内质网中未折叠或错误折叠的蛋白增多时,应激信号就通过内质网膜传递到细胞核中,进而引起的一系列特定的靶基因转录上调和蛋白质翻译水平下调,使细胞能继续存活。持续的内质网应激和UPR都可以激活内质网自噬,内质网应激、UPR和内质网自噬都是内质网的主要反应,参与调控内质网结构与功能。
调控内质网功能平衡的一个主要机制就是UPR。内质网中大量未折叠蛋白和错误折叠蛋白累积,而引起内质网损伤,形成内质网应激,激活细胞的UPR。涉及UPR的内质网膜上应激传感蛋白有3个,分别是肌醇依赖酶1α(inositol-requiring enzyme 1,IRE-1α)、激活转录因子6(activating transcription factor 6,ATF6)和蛋白激酶样内质网激酶(PKR-like ER kinase,PERK)。当UPR在起始阶段时,内质网膜上的应激传感蛋白被激活,减少或降解未折叠的蛋白并改善蛋白折叠功能[20]。在某些情况下,内质网的这种蛋白修复功能可以因突变或特殊环境条件而改变。可通过特定突变蛋白的表达如抗胰蛋白酶,和特殊化学试剂如DTT的处理来构建UPR模型[21],这种模型可引起内质网中蛋白发生错误折叠和蛋白聚集物的积累。有研究发现在UPR模型的细胞中,自噬参与错误折叠蛋白和聚集蛋白的清除过程[22-23]。其不仅参与了细胞中多条信号通路,也是细胞中重要的应激反应机制[24]。通过光学显微镜对应激-自噬的研究表明,真核生物中内质网应激可以诱导自噬体的形成[25],但是文中并未提到自噬类型。在哺乳动物细胞中,鞘氨醇-1-磷酸(S1P)的磷酸酶减少引起的内源性S1P的水平增加会导致内质网应激触发自噬[26]。该诱导过程是哺乳动物雷帕霉素靶蛋白(mammalian target of rapamycin,mTOR)非依赖性的和PERK、IRE1和ATF6依赖性的。内质网应激导致Ca2+从内质网释放到细胞质中启动各种信号通路,其中有一些可能参与诱导自噬[27-28]。在内质网应激过程中,自噬抑制剂或敲除ATG蛋白都可以阻断自噬体的形成[21]。另有报道显示UPR激活可诱导自噬[8],引起自噬体的增多[29-30]。
Bernales等[2]发现在酵母细胞中,激活UPR可以引起内质网重构,发生重构的内质网被双层膜结构包裹并扩展成囊泡,该囊泡中只包含内质网膜结构,不含其他细胞器及胞质。 采用内质网标志蛋白的免疫标记显示被吞噬的囊泡膜,提示其来自内质网,而囊泡的边界膜上常附着核糖体,由此推测被吞噬的囊泡和边界膜均来源于内质网,据此提出这种选择性自噬为内质网自噬。通过电子显微镜观察可以发现UPR引起的自噬体常含有多层膜结构,该现象在饥饿诱导的自噬体中并没出现,多层膜结构提示UPR引起的自噬可能是内质网自噬。当UPR无法改善内质网功能时,细胞就会启动内质网自噬。内质网自噬可以清除损伤的内质网和错误加工的蛋白,展示出对UPR及内质网的降解作用,以维持细胞内稳态[31]。Hamasaki等[15]发现在抑制非选择性自噬的酵母中,饥饿可以诱导自噬体特异性吞噬内质网,发生内质网自噬。Kamimoto等[16]发现在哺乳动物肝脏细胞中,α1-抗胰蛋白酶Z突变体会聚集在内质网,并通过内质网自噬途径清除聚集体。Rubio等[17]研究发现,在鼠纤维肉瘤细胞中,光损伤可以引起细胞发生氧化应激,进而诱导内质网损伤并发生内质网自噬。
Hamasaki等[11]的结果显示饥饿处理后,内质网与线粒体的连接位点上的自噬体标志物ATG14,偶联内质网蛋白syntaxin 17,调节内质网自噬,因此syntaxin 17 /ATG14可能可以作为内质网自噬的标志蛋白。在酵母中的Rab蛋白Ypt(yeast protein transport)可能对选择性自噬具有调节作用[32]。Ypt1负责调节内质网向高尔基体转运,也可能调控内质网转化成自噬体,控制内质网自噬体的发生过程[12]。
内质网自噬有双重作用,一是在蛋白折叠应激过程中,部分内质网被损坏或是含有大量不能通过其他方式处理的错误折叠蛋白,内质网自噬体的形成有利于隔离这部分无法发挥正常功能的内质网;二是在蛋白折叠应激减弱时,内质网自噬可促进内质网形状恢复正常。
4 内质网自噬参与或抑制细胞凋亡
内质网自噬作为调节内质网功能的方式之一,在一定程度上可以清除内质网损伤引起的细胞反应,如内质网应激和UPR,甚至是细胞凋亡等。但如果内质网应激过于强烈或持久,超过了细胞负荷时,UPR和内质网自噬不足以改善内质网功能,维持内质网内稳态时,细胞就会启动由内质网应激所介导的C/EBP同源蛋白(C/EBP homologous protein,CHOP)、c-Jun氨基酸末端激酶(c-Jun N-terminalkinase,JNK)或半胱氨酸天冬氨酸特异性蛋白酶(caspase)通路,引起细胞凋亡(Fig 2)。不同程度的内质网应激可引发不同程度的细胞反应,凋亡和自噬是细胞中重要且相互联系的反应机制。过度的内质网应激会通过IRE1α和PERK通路共同作用,导致Bip和CHOP上调,CHOP是UPR3个感受蛋白IRE-1α、ATF和PERK的共同靶基因,可以调节Bcl-2蛋白家族成员的表达水平,促进Bim(bcl-2 interacting mediator of cell death) 转录而抑制Bcl-2的表达,上调BH3蛋白(BH3-only proteins)的转录,活化Bax/Bak,促使细胞凋亡[24]。未折叠蛋白引起内质网应激,激活未折叠反应,通过pERK使eIF2a磷酸化,进而降低整个细胞蛋白的合成,而激活转录因子4(activating transcription factor 4, ATF4)mRNA的转录增多。ATF4作为氧化还原平衡、氨基酸代谢、蛋白折叠、自噬和凋亡等综合应激反应的调节剂。内质网应激过程中自噬的上调是参与细胞自救反应,内质网自噬可以清除未折叠的蛋白、蛋白聚集体和受损的细胞器。因此,ATF4可以引起自噬,被认为是细胞应激过程中的保护因子[33],但是ATF4也可以使CHOP的转录增加,下调Bcl-2,引起细胞凋亡(Fig 2)。内质网应激过程通过UPR信号传递,激活细胞的内质网自噬功能,清除内质网中的未折叠蛋白、蛋白聚集体和受损的内质网,以维持内质网稳态,使细胞存活。细胞内质网稳态被严重破坏,内质网自噬无法改善内质网状态时,凋亡信号就会被激活,诱导细胞凋亡。在内质网自噬和凋亡信号通路中,CHOP与ATF4可能作为重要的调节信号分子,决定细胞的最终走向。Rubio等[17]研究发现,内质网氧化损伤可以引起内质网自噬和细胞凋亡。内质网氧化损伤后发生内质网自噬,并传递ROS到附近的线粒体,引发线粒体损伤。线粒体损伤使细胞内氧化信号放大,激活caspase-3,引起细胞凋亡。由此可见,内质网自噬对细胞凋亡有双重作用,既可以拮抗细胞凋亡,又参与细胞凋亡。虽然自噬与凋亡的研究已经很多,但是内质网自噬与凋亡之间的关系仍有很多未知,需要进一步研究。
Tab 1 Tab 1 The differences between autophagy and selective autophagy
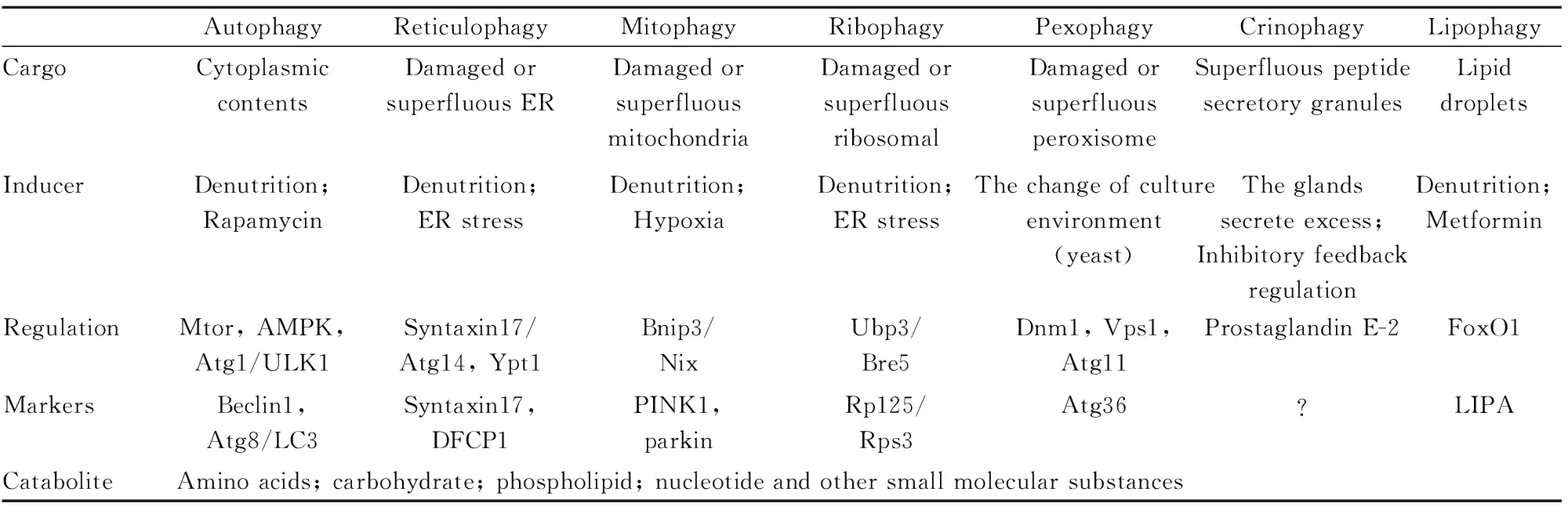
AutophagyReticulophagyMitophagyRibophagyPexophagyCrinophagyLipophagyCargoCytoplasmiccontentsDamagedorsuperfluousERDamagedorsuperfluousmitochondriaDamagedorsuperfluousribosomalDamagedorsuperfluousperoxisomeSuperfluouspeptidesecretorygranulesLipiddropletsInducerDenutrition;RapamycinDenutrition;ERstressDenutrition;HypoxiaDenutrition;ERstressThechangeofcultureenvironment(yeast)Theglandssecreteexcess;InhibitoryfeedbackregulationDenutrition;MetforminRegulationMtor,AMPK,Atg1/ULK1Syntaxin17/Atg14,Ypt1Bnip3/NixUbp3/Bre5Dnm1,Vps1,Atg11ProstaglandinE-2FoxO1MarkersBeclin1,Atg8/LC3Syntaxin17,DFCP1PINK1,parkinRp125/Rps3Atg36?LIPACataboliteAminoacids;carbohydrate;phospholipid;nucleotideandothersmallmolecularsubstances
ER: endoplasmic reticulum;LIPA: lysosomal acid lipase A
5 内质网自噬与其他选择性自噬的关联
内质网自噬的形成是一个复杂的过程,其常常伴随核糖体自噬和线粒体自噬过程一起发生或相互传递信号,共同在细胞中发挥作用。内质网自噬和核糖体自噬同样在细胞中发挥作用,参与调节细胞内蛋白的合成和代谢。当细胞发生内质网自噬时,内质网会传递信号到线粒体,诱导线粒体发生选择性自噬。Tab 1中比较了内质网自噬与自噬及其他几种选择性自噬间的差异。
5.1 核糖体自噬核糖体在蛋白质形成过程中具有重要的作用,主要负责细胞mRNA的翻译和蛋白的合成。在自噬的研究中,核糖体被当做细胞质内一部分被自噬体吞噬,并且作为自噬体中细胞质的标记物[3]。最近研究表明,营养缺乏使酵母中核糖体迅速被自噬体选择性吞噬降解,即“核糖体自噬(ribophagy)”[3]。核糖体蛋白125 (ribosomal protein 125,RP125) 和核糖体蛋白s3水平可以作为核糖体自噬的标志蛋白。 核糖体自噬的降解过程需要泛素蛋白酶复合物Ubp3/Bre5的催化活性,在Ubp3和Bre5敲除的细胞中营养缺乏可促进核糖体积累,使核糖体代谢异常,引起细胞凋亡[3]。核糖体的生成和蛋白的翻译是细胞重要的能量代谢过程,因此当营养充足的条件下,真核细胞中有大量稳定的成熟核糖体来翻译细胞所需的蛋白[34]。当细胞环境改变,如营养缺乏时,细胞会通过核糖体自噬降解多余的核糖体,减慢蛋白翻译的速度。核糖体中含有大量蛋白复合体,因此,核糖体自噬可以为细胞提供大量生存所必需的氨基酸等代谢产物和能量,维持细胞稳态。与内质网自噬降解功能丧失或受损的内质网的作用类似,核糖体自噬也可以清除细胞内无效的与组装错误的核糖体,避免蛋白翻译错误[31]。
5.2 内质网自噬可诱导线粒体自噬线粒体作为细胞的能量工厂,为细胞提供能量和生物合成所需的底物,决定着细胞的命运。细胞异常代谢过程中常会诱发线粒体损伤,进而通过一系列的蛋白信号引发细胞凋亡。为了维持细胞功能,受损和多余的线粒体可通过选择性自噬途径被溶酶体降解,该过程被称为“线粒体自噬”[33, 35]。当细胞中线粒体损伤时,线粒体自噬作为一种调控线粒体质量的调控机制被激活,维持细胞稳态。线粒体损伤的严重程度及引发线粒体损伤的因素的强弱共同决定了细胞的命运,如果线粒体自噬及时清除细胞内受损的线粒体,可以保护细胞不受异常线粒体代谢和促凋亡信号蛋白的危害。线粒体自噬的直接受体促凋亡线粒体蛋白Bnip3(BCL2 and 19-kDa interacting protein-3)和Nix(Bnip3L)与LC3蛋白相互作用,并募集LC3到损伤的线粒体上引起线粒体自噬的发生[36-37]。E3泛素连接酶Parkin和丝氨酸/苏氨酸激酶Pink l参与了线粒体膜电位降低引起的线粒体自噬的发生[38],是线粒体自噬的标志蛋白。
内质网自噬与线粒体自噬具有紧密的联系。Noemí Rubio等[17]通过对内质网光损伤引发的自噬的研究发现,当内质网发生光损伤6 h之后,自噬体包含的内质网的数量明显高于所包含的线粒体的数量,而线粒体的形态发生变化,成为网状结构。当光损伤16 h后,自噬体所包含的线粒体的数量却远高于内质网的数量。Noemí Rubio等研究提示当内质网发生光氧化应激早期,内质网自噬为主要的自噬方式,随着内质网自噬的增强,内质网传递磷脂氢过氧化物到附近的线粒体并引起线粒体损伤,激活线粒体自噬,因此到光氧化应激后期线粒体自噬是主要的自噬方式。
6 内质网自噬参与疾病的发生发展
内质网自噬是参与调控内质网功能的主要方式之一,可以有效地清除细胞中受损的内质网和无效蛋白质,从而预防疾病的发生。但在一些病理情况下,内质网自噬水平也会发生改变,过度激活的内质网自噬可过度清除细胞内正常内质网,或通过传导信号激活其他细胞反应,反而使机体病变加剧。以下针对内质网自噬在胰岛素抵抗、帕金森病和肾脏疾病中的作用,论述内质网自噬与这些疾病的关系。抑制或激活细胞内质网自噬成为潜在的预防和治疗内质网相关疾病的新靶点。
6.1 内质网自噬参与胰岛素抵抗的形成胰岛素抵抗(insulin resistance)是肥胖相关的代谢综合征和2型糖尿病的一个标志,而内质网可能在胰岛素抵抗的发生发展过程中具有重要作用。在病理情况下,营养和能量状态改变如肥胖会使内质网超负荷工作,导致错误或未折叠蛋白的堆积,引发内质网应激。内质网应激会使分子伴侣葡萄糖调节蛋白78(glucose-regulated protein 78)从内质网膜上的PERK、IRE1和ATF6上脱离调节糖代谢,PERK、IRE1和ATF6属于内质网膜上的膜信号传感器,属于细胞对内质网发生未折叠反应做出的应答[39]。关于内质网应激的研究范围已经拓展到胰脏β细胞、肝细胞和脂肪细胞[40]。蛋白酪氨酸磷酸酶1B(protein-tyrosine phosphatase 1B,PTP1B)位于内质网上,参与胰岛素信号转导的负调节。PTP1B可以增强IRE-1α介导的信号通路上调UPR。内质网应激上调PTP1B使细胞的葡萄糖摄取功能受损[41],该过程涉及多个信号通路,包括UPR相关的ATF6的激活,与转录因子YB-1和NF-κB (nuclear factor kappa-light-chain-enhancer of activated B cells) 偶联。高脂饮食可以激活UPR,引发内质网应激[42],不同程度的内质网应激可引发不同程度的细胞反应,如应激的起始阶段会引起内质网自噬来清除细胞中受损的内质网和聚集的蛋白质,维持细胞器功能。干预内质网自噬的发生可以有效清除细胞内损伤的内质网和聚集的蛋白质,减少内质网应激的发生,进而维护细胞葡萄糖摄取功能,因此,干预内质网自噬可能成为治疗胰岛素抵抗相关疾病,如代谢综合征和2型糖尿病的新方法。
6.2 内质网自噬参与帕金森病的发生发展帕金森病(Parkinsonism)与内质网自噬有密切联系[43]。帕金森病中α-synuclein异常折叠蛋白增多以及多巴胺能神经元减少均会引发内质网应激[44]。内质网应激可以通过UPR将未折叠和错误折叠蛋白进行正确折叠,而过度的内质网应激会导致内质网损伤引起神经细胞的自噬和凋亡。在细胞和动物模型中,自噬参与帕金森病的病理过程,α-synuclein的异常折叠无法通过蛋白酶体的桶状结构,也无法被蛋白酶体降解,从而集聚在内质网中使内质网损伤,引发内质网应激和自噬,以清除损伤的内质网[45]。但是Zeng等[46]研究发现抑制内质网应激可以在帕金森病中起到神经保护作用。在帕金森病的发生过程中,内质网自噬可能作为一种神经细胞保护机制,清除神经细胞内受损的内质网,以维护神经细胞的稳态,防止帕金森病的发生,而在帕金森病的发展过程中,神经细胞内稳态严重失衡,可能过度激活内质网自噬,使神经细胞进一步受损。利用该机制,激活或抑制内质网自噬可能为预防或治疗帕金森病提供了一个新的方向。
6.3 内质网自噬是治疗肾脏疾病的新靶点肾病蛋白nephrin是肾小球裂孔膜上的跨膜蛋白,要经过内质网的加工修饰才转至细胞膜上,在维持肾小球选择通透性及正常功能中起关键作用。nephrin的突变会导致蛋白尿或先天性肾病综合征的发生。Drozdova等[47]对人体nephrin的错义突变体进行研究发现,突变体表现出糖基化受损和增强了突变体与内质网分子伴侣和钙联接蛋白的结合,并且nephrin突变体在内质网聚集,激活UPR的转录因子-6(TF-6)信号通路,并增强内质网分子伴侣表达。同时nephrin突变体增强细胞的泛素化作用,发生内质网自噬,减少突变体进入质膜。蛋白尿或先天性肾病综合征等肾脏疾病中,增强内质网自噬可能会有效清除病理细胞中错误折叠和聚集的蛋白质及受损的内质网,起到治疗疾病的作用。因此内质网自噬可能成为治疗蛋白尿或先天性肾病综合征等肾脏疾病的新靶点。
7 展望
细胞通过内质网自噬清除内部损伤、失效及多余的胞质组分,维持细胞内环境的稳定。内质网在多种疾病的发生发展中有重要作用,众多机体病变是因为内质网的功能缺陷或丧失引起的,如上文提到的胰岛素抵抗、肾脏疾病及神经退行性疾病等。众多因素可以引起内质网内稳态的改变,使内质网功能缺陷或丧失,引发细胞应激反应,激活内质网自噬。内质网自噬是当前研究的热点,但是仍有很多问题没有解决。首先就是自噬体的起源问题,自噬体的双层膜结构可能是有受损并发生结构重构的内质网提供的;其次,自噬体与溶酶体融合降解的调控过程仍然未知。哺乳动物中,自噬体与溶酶体融合前可能先与核内体融合,包含内质网的自噬体可以明显地存储在细胞质,而酵母中自噬体可以直接传递到液泡;还有,内质网自噬过程中涉及到很多已知的ATG蛋白,内质网自噬和自噬之间的联系与区别仍不是很明确,自噬的诱发条件比内质网自噬的严峻,因此内质网自噬可能只是细胞发生的局部自噬过程,或内质网自噬是细胞自噬发生的初级阶段;最后,内质网自噬的形成过程,内质网靶点结构位置的重构,核糖体脱落,自噬体包裹内质网,内质网膜的折叠或部分膜断裂,这些过程的调节和具体的作用方式的探索在内质网自噬的研究具有重要的意义。内质网自噬和内质网应激过程都有多种转录因子和激酶调控,这些位点可能成为预防和治疗内质网相关疾病的药物靶点。内质网自噬的进一步研究不仅可以为细胞中应激-反应的研究提供新的思路,还可以为疾病的预防和治疗提供新的方向。
[1] 林 超,刘兆国,钱 星,等. 自噬在心血管疾病中的作用研究进展[J]. 中国药理学通报, 2014, 30(10): 1347.
[1] Lin C, Liu Z G, Qian X, et al. Research progress on the role of autophagy in cardiovascular diseases[J].ChinPharmacolBull, 2014,30(10):1347
[2] Bernales S, Schuck S, Walter P. ER-phagy: selective autophagy of the endoplasmic reticulum [J].Autophagy, 2007, 3(3): 285-7.
[3] Kraft C, Deplazes A, Sohrmann M, et al. Mature ribosomes are selectively degraded upon starvation by an autophagy pathway requiring the Ubp3p/Bre5p ubiquitin protease [J].NatureCellBiol, 2008, 10(5): 602-10.
[4] Lemasters J J. Selective mitochondrial autophagy, or mitophagy, as a targeted defense against oxidative stress, mitochondrial dysfunction, and aging [J].RejuvenationRes, 2005, 8(1): 3-5.
[5] Smith R E, Farquhar M G. Lysosome function in the regulation of the secretory process in cells of the anterior pituitary gland [J].JCellBiol, 1966, 31(2): 319-47.
[6] Tuttle D L, Lewin A S, Dunn W A. Selective autophagy of peroxisomes in methylotrophic yeasts [J].EurJCellBiol, 1993, 60(2): 283-90.
[7] Singh R, Kaushik S, Wang Y, et al. Autophagy regulates lipid metabolism [J].Nature, 2009, 458(7242): 1131-5.
[8] Yorimitsu T, Nair U, Yang Z, et al. Endoplasmic reticulum stress triggers autophagy [J].JBiologicalChem, 2006, 281(40): 30299-304.
[9] Rabinowitz J D, White E. Autophagy and metabolism [J].Science, 2010, 330(6009): 1344-8.
[10] Tooze S A, Yoshimori T. The origin of the autophagosomal membrane [J].NatureCellBiol, 2010, 12(9): 831-5.
[11] Hamasaki M, Furuta N, Matsuda A, et al. Autophagosomes form at ER-mitochondria contact sites [J].Nature, 2013, 495(7441): 389-93.
[12] Lipatova Z, Shah A H, Kim J J, et al. Regulation of ER-phagy by a Ypt/Rab GTPase module [J].MolBiolCell, 2013, 24(19): 3133-44.
[13] Axe E L, Walker S A, Manifava M, et al. Autophagosome formation from membrane compartments enriched in phosphatidylinositol 3-phosphate and dynamically connected to the endoplasmic reticulum [J].JCellBiol, 2008, 182(4): 685-701.
[14] Klionsky D J, Abdalla F C, Abeliovich H, et al. Guidelines for the use and interpretation of assays for monitoring autophagy [J].Autophagy, 2012, 8(4): 445-544.
[15] Hamasaki M, Noda T, Baba M, et al. Starvation triggers the delivery of the endoplasmic reticulum to the vacuole via autophagy in yeast [J].Traffic, 2005, 6(1): 56-65.
[16] Kamimoto T, Shoji S, Hidvegi T, et al. Intracellular inclusions containing mutant alpha1-antitrypsin Z are propagated in the absence of autophagic activity [J].JBiologicalChem, 2006, 281(7): 4467-76.
[17] Rubio N, Coupienne I, Di Valentin E, et al. Spatiotemporal autophagic degradation of oxidatively damaged organelles after photodynamic stress is amplified by mitochondrial reactive oxygen species [J].Autophagy, 2012, 8(9):1312-24.
[18] Pearson G L, Mellett N, Chu K Y, et al. Lysosomal acid lipase and lipophagy are constitutive negative regulators of glucose-stimulated insulin secretion from pancreatic beta cells [J].Diabetologia, 2014, 57(1): 129-39.
[19] 刘春蕾,何昆仑,王莉莉. 基于内质网应激途径的细胞保护策略的研究进展 [J]. 中国药理学通报, 2011, 27(4): 455-8.
[19] Liu C L, He K L, Wang L L. Research of cell protection based on ER stress[J].ChinPharmacolBull, 2011, 27(4): 455-8.
[20] Braakman I, Bulleid N J. Protein folding and modification in the mammalian endoplasmic reticulum [J].AnnualReviewBiochem, 2011, 80: 71-99.
[21] Fujita E, Kouroku Y, Isoai A, et al. Two endoplasmic reticulum-associated degradation (ERAD) systems for the novel variant of the mutant dysferlin: ubiquitin/proteasome ERAD(I) and autophagy/lysosome ERAD(Ⅱ) [J].HumanMolgenetics, 2007, 16(6): 618-29.
[22] Kruse K B, Brodsky J L, Mccracken A A. Characterization of an ERAD gene as VPS30/ATG6 reveals two alternative and functionally distinct protein quality control pathways: one for soluble Z variant of human alpha-1 proteinase inhibitor (A1PiZ) and another for aggregates of A1PiZ [J].MolBiologyCell, 2006, 17(1): 203-12.
[23] Kruse K B, Dear A, Kaltenbrun E R, et al. Mutant fibrinogen cleared from the endoplasmic reticulum via endoplasmic reticulum-associated protein degradation and autophagy: an explanation for liver disease [J].AmJPathol, 2006, 168(4): 1299-308.
[24] Matsumoto H, Miyazaki S, Matsuyama S, et al. Selection of autophagy or apoptosis in cells exposed to ER-stress depends on ATF4 expression pattern with or without CHOP expression [J].BiolOpen, 2013, 2(10): 1084-90.
[25] Kouroku Y, Fujita E, Tanida I, et al. ER stress (PERK/eIF2alpha phosphorylation) mediates the polyglutamine-induced LC3 conversion, an essential step for autophagy formation [J].CellDeathDifferent, 2007, 14(2): 230-9.
[26] Lepine S, Allegood J C, Park M, et al. Sphingosine-1-phosphate phosphohydrolase-1 regulates ER stress-induced autophagy [J].CellDeathDifferent, 2011, 18(2): 350-61.
[27] Hoyer-Hansen M, Bastholm L, Szyniarowski P, et al. Control of macroautophagy by calcium, calmodulin-dependent kinase kinase-beta, and Bcl-2 [J].MolCell, 2007, 25(2): 193-205.
[28] Hoyer-Hansen M, Jaattela M. Connecting endoplasmic reticulum stress to autophagy by unfolded protein response and calcium [J].CellDeathDifferent, 2007, 14(9): 1576-82.
[29] Ogata M, Hino S, Saito A, et al. Autophagy is activated for cell survival after endoplasmic reticulum stress [J].MolCellularBiol, 2006, 26(24): 9220-31.
[30] Ding W X, Ni H M, Gao W, et al. Differential effects of endoplasmic reticulum stress-induced autophagy on cell survival [J].JBiologicalChem, 2007, 282(7): 4702-10.
[31] Cebollero E, Reggiori F, Kraft C. Reticulophagy and ribophagy: regulated degradation of protein production factories [J].InternatJCellBiol, 2012, 2012: 182834.
[32] Lipatova Z, Belogortseva N, Zhang X Q, et al. Regulation of selective autophagy onset by a Ypt/Rab GTPase module [J].ProceedingsNationalAcadSciUSA, 2012, 109(18): 6981-6.
[33] Ameri K, Harris A L. Activating transcription factor 4 [J].InternatJBiochemCellBiol, 2008, 40(1):14-21.
[34] Warner J R. The economics of ribosome biosynthesis in yeast [J].TrendsBiochemSci, 1999, 24(11): 437-40.
[35] Kim I, Rodriguez-Enriquez S, Lemasters J J. Selective degradation of mitochondria by mitophagy [J].ArchBiochemBiophysics, 2007, 462(2): 245-53.
[36] Hanna R A, Quinsay M N, Orogo A M, et al. Microtubule-associated protein 1 light chain 3 (LC3) interacts with Bnip3 protein to selectively remove endoplasmic reticulum and mitochondria via autophagy [J].JBiologChem, 2012, 287(23): 19094-104.
[37] Novak I, Kirkin V, Mcewan D G, et al. Nix is a selective autophagy receptor for mitochondrial clearance [J].EMBOReports, 2010, 11(1): 45-51.
[38] Vives-Bauza C, Zhou C, Huang Y, et al. PINK1-dependent recruitment of Parkin to mitochondria in mitophagy [J].ProcNationalAcadSciUSA, 2010, 107(1): 378-83.
[39] Malhotra J D, Kaufman R J. Endoplasmic reticulum stress and oxidative stress: a vicious cycle or a double-edged sword? [J].AntioxidantsRedoxSignal, 2007, 9(12): 2277-93.
[40] Fu S, Watkins S M, Hotamisligil G S. The role of endoplasmic reticulum in hepatic lipid homeostasis and stress signaling [J].CellMetabolism, 2012, 15(5): 623-34.
[41] Panzhinskiy E, Hua Y, Culver B, et al. Endoplasmic reticulum stress upregulates protein tyrosine phosphatase 1B and impairs glucose uptake in cultured myotubes [J].Diabetologia, 2013, 56(3): 598-607.
[42] Panzhinskiy E, Ren J, Nair S. Protein tyrosine phosphatase 1B and insulin resistance: Role of endoplasmic reticulum stress/reactive oxygen species/nuclear factor kappa B axis [J].PloSOne, 2013, 8(10): e77228.
[43] Matus S, Castillo K, Hetz C. Hormesis: protecting neurons against cellular stress in Parkinson disease [J].Autophagy, 2012, 8(6): 997-1001.
[44] Chung C Y, Khurana V, Auluck P K, et al. Identification and rescue of alpha-synuclein toxicity in Parkinson patient-derived neurons [J].Science, 2013, 342(6161): 983-7.
[45] Ghavami S, Shojaei S, Yeganeh B, et al. Autophagy and apoptosis dysfunction in neurodegenerative disorders [J].ProgNeuroBiol, 2013, 112: 24-49.
[46] Zeng X S, Jia J J, Kwon Y, et al. The role of thioredoxin-1 in suppression of endoplasmic reticulum stress in Parkinson disease [J].FreeRadicalBiologyMed, 2013, 67: 10-8.
[47] Drozdova T, Papillon J, Cybulsky A V. Nephrin missense mutations: induction of endoplasmic reticulum stress and cell surface rescue by reduction in chaperone interactions [J].PhysiolReports, 2013, 1(4): e00086.
Reticulophagy-a new target for diseases prevention and treatment
LI He-ning, LI Lan-fang, CHEN Lin-xi
(InstituteofPharmacyandPharmacology,UniversityofSouthChina,HengyangHunan421001,China)
The cells activate selective autophagy and remove damaged endoplasmic reticulum or fragments in cells when changes the function of endoplasmic reticulum, known as the endoplasmic reticulum autophagy. Reticulophagy is a selective autophagy and can remove damaged endoplasmic reticulum or fragments in cells. The function of reticulophagy is to improve the inside environment of cells and protect the cells. As a new field of selective autophagy research, it and the other types of selective autophagy are closely interconnected. Meanwhile, it is also associated with cell apoptosis and has a very important influence on diseases. The paper reviews the formation of reticulophagy, intracellular regulation and the interaction between other selective autophagys. In addition, we also describe the close relationship between reticulophagy and some diseases, such as diabetes, neurodegenerative diseases and renal diseases. Hence, reticulophagy gradually becomes a new target for disease prevention and treatment.
reticulophagy; autophagy; unfolded protein response; endoplasmic reticulum stress; selective autophagy; medical targets
时间:2015-3-3 11:08 网络出版地址:http://www.cnki.net/kcms/detail/34.1086.R.20150303.1108.002.html
2014-11-19,
2014-12-21
国家自然科学基金资助项目(No 81270420,81470434);湖南省自然科学基金省市联合(衡阳)基金重点项目(No 12JJ8013);湖南省自然科学基金资助项目(No 14JJ3102);国家博士后基金(No 2014M560647);湖南省“十二五”重点学科建设项目资助
李赫宁(1989-),男,硕士生,研究方向:分子药理学,E-mail: 285035163@qq.com; 陈临溪(1965-),男,博士,教授,博士生导师,主要研究方向:分子药理学与新药分子设计评价,通讯作者,Tel:0734-8282614, E-mail: lxchen6@126.com
10.3969/j.issn.1001-1978.2015.03.002
A
1001-1978(2015)03-0302-07
R-05;R329.24;R587.1;R692;R745.7
