一个腓骨肌萎缩症2型核心家系临床表型及矫形手术治疗
2021-01-07段丽芬刘红仙杨昭庆王惠萍陈文洋
段丽芬,刘红仙,杨昭庆,王惠萍,陈文洋
腓骨肌萎缩症(Charcot-Marie-Tooth disease,CMT),又称遗传性运动感觉神经病(hereditary motor and sensory neuropathy,HMSN),由法国神经病学家Charcot 和Marie以及英国神经病学家Tooth于1886年率先报告。CMT是一组具有高度临床和遗传异质性的周围神经单基因遗传病[1],其遗传方式有:常染色体显性遗传、常染色体隐性遗传和X连锁显性和隐性遗传[2,3],目前发现80余种致病基因,近1000余个突变可导致该病的发生,CMT发病率约为1/2500到1/10000不等。CMT通常于儿童期或青少年期发病,临床主要表现为慢性进行性四肢远端肌无力和肌萎缩伴随感觉减退、腱反射消失或减退,伴高弓足和脊柱侧弯等骨骼畸形为特征的周围神经病。CMT目前尚无逆转病程的特异性治疗方法,可穿戴矫形鞋联合物理治疗、外科矫形手术等对症治疗,手术外科治疗的报道较少。基于对CMT的遗传学和临床诊治的现状,本研究对一个MFN2基因确诊的CMT家系进行临床特征和矫形手术治疗效果进行总结分析,以期对临床治疗进行指导。
1 临床资料
一个来自云南省曲靖山区的核心家系,包括先证者及其父亲、母亲和妹妹(见图1)。先证者,男,10岁,3岁前里程碑发育正常,3岁后出现双足姿势异常,行走时双足内收、无力,易摔倒,逐渐加重后出现双足完全内翻,双侧足背外侧着地行走,双下肢肌肉萎缩,5岁后出现双手掌肌肉萎缩、姿势异常(手指屈曲,不能伸直),写字困难,无法进行精细动作。个人史:患儿为G1P1,足月顺产,出生体重2.5 Kg,出生时有窒息史。既往史:否认药物过敏史,否认外伤手术史;家族史:患儿母亲8~9岁逐渐出现行走姿势异常及四肢肌肉萎缩,之后出现下肢瘫痪,现仅能独坐,不能站立和行走,四肢纤细、消瘦,肌容积明显减少;语言和智力发育正常。G2P2:女,8岁,4岁后出现与先证者类似症状,下肢无力姿势异常后出现双足内翻,足背外侧行走,肌肉萎缩无力。查体:语言认知正常,双手指屈曲无法伸直,双下肢马蹄内翻足,足内侧可见皮肤皱褶,足外侧皮质增厚、胼胝形成,双侧足底不能着地,双侧足背外侧着地行走,双侧跟腱及跖腱膜紧、挛缩,双足背伸、屈曲、外展、内收功能均受限。颅神经查体未见异常,四肢肌肉萎缩,肌容积明显减低,远端肌肉萎缩明显,四肢肌力Ⅳ级,四肢肌张力增高,腹壁反射对称引出,膝反射未引出,双侧Babinski征未引出,脑膜刺激征阴性。
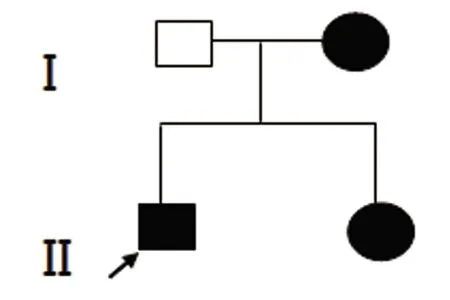
图1 腓骨肌萎缩综合征患者家系图谱,箭头指示先证者
辅助检查:血生化、铜蓝蛋白、血常规、血浆氨、血气、脑脊液常规和生化、甲状腺功能、肾上腺功能、TORCH、抗核抗体、微量元素均正常。周围神经传导功能:四肢多条受检神经运动、感觉传导功能异常,表现为:(1)双侧胫、腓总、正中、尺神经复合肌肉动作电位(CMAP)均未引出;双侧股、桡神经CMAP波幅降低;(2)各受检神经F波均未引出;(3)各受检神经感觉神经动作电位(SNAP)均未引出。神经传导检测结果提示有四肢广泛性周围神经病变,传导阻滞,轴索病变为主可能,远端重于近端。双髋关节未见明显异常。肝胆胰脾肾超声正常,心脏超声正常,头部和腰骶椎MRI正常。基因全外显子测序结果:在先证者、母亲和妹妹3者MFN2 基因发现c.1090C>T(编码区第1090号核苷酸由C变为T)的杂合核苷酸变异,该变异导致第364号氨基酸由Arg变为Trp(p.Arg364Trp),为错义变异。患儿父亲基因位点无突变。该变异可能导致蛋白质功能受到影响,此变异的致病性已经有文献报道。
治疗经过及结局:基因结果未回报前为排外多巴反应性肌张力障碍予以小剂量美多芭实验性口服治疗一个月,患儿症状无缓解。基因回报确诊CMT2型后完善术前评估检查无明显禁忌证,给予手术治疗。手术方式为:双侧跟腱、趾腱膜松解+马蹄内翻足挛缩关节松解牵伸外固定架固定矫形术。术中松解跟腱及趾腱膜,手法松解踝足关节,复位距舟关节,足踝畸形改善,小腿周围及踝足周围连接Ilizarov骨科外固定支架踝关节架。通过微创小切口逐步牵伸矫形的方式,使患者足底可以着地正常行走,减缓肌肉萎缩,达到手术治疗的目的。现治疗8个月,外固定支架已取,目前恢复情况可,可自行下地行走,无不适症状,畸形无反复(见图2、图3)。其母亲因发病时间较长、肢体和躯干畸形严重暂无法进行进一步手术干预(见图4)。
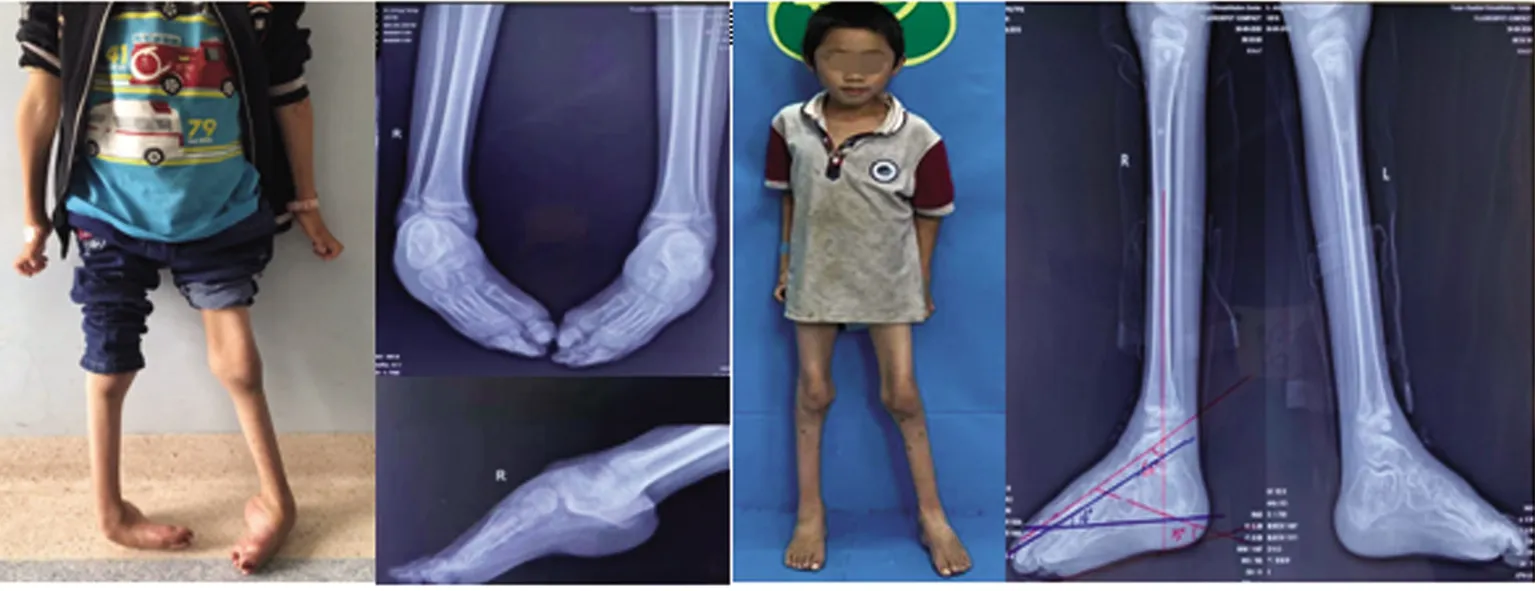
图2 先证者手术前(图A、B)后(图C、D)临床特征和下肢X片
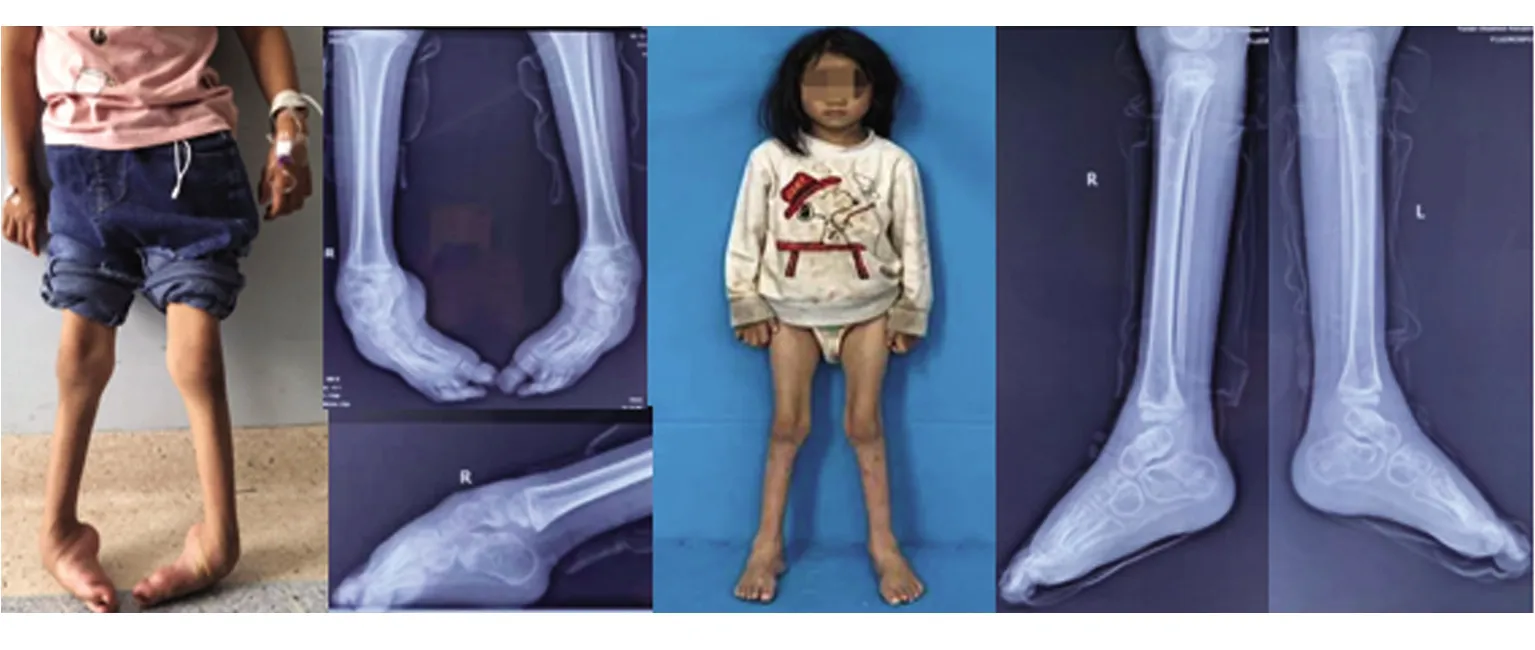
图3 先证者妹妹手术前(图A、B)后(图C、D)临床特征和下肢X片
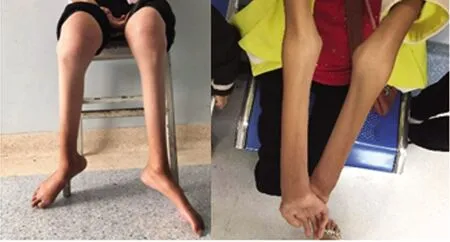
图4 先证者母亲
2 讨 论
CMT是一类包括损害周围神经的遗传性感觉和运动神经疾病。CMT儿童期或青少年期起病,2/3患儿10岁前发病,起病隐袭,腓肠神经最先受累,肌无力和肌萎缩先出现在肢体远端,之后逐渐累及上肢及四肢近端,出现肢体畸形影响四肢的运动功能,伴随感觉减退、腱反射消失或减退,临床常出现高足弓、跨阈步态、足下垂、足内翻、锤状趾、鹤腿征、倒置的酒瓶样畸形、爪型手或猿手、脊柱侧弯等身体严重畸形[4,5]。颅神经和括约肌功能不受影响,大部分患者智力正常。病程终生慢性进行性加重,多数患者疾病进展缓慢,出现轻至中度功能损害,但不影响预期寿命,少数患者病情进展迅速出现呼吸肌无力。
CMT可分为:CMT1、CMT2、CMT3、CMT4、CMTDI、CMTRI、CMTX等型,根据遗传方式,CMT可表现为常染色体显性、隐性遗传以及X-连锁显性和隐性遗传,其中以常染色体显性遗传多见。CMT2又包括了CMT2A、CMT2B和CMT2D等[6]。CMT2主要表现为神经传导波幅降低,传导速度正常或轻度减慢,正中神经运动传导速度>38 m/s。神经活检为慢性轴索变性和再生,有髓纤维减少,神经再生簇形成。CMT2的致病基因有:MFN2、KIF1B、RAB7、LMNA等。MFN2是CMT2A中最主要的致病基因。MFN2是编码线粒体GTP酶的核基因,对于线粒体的结构完整性、形态和运输至关重要[7]。通过影响氧化磷酸化而参与能量代谢[8]。已发现MFN2的变异约有190多种,多为常染色体显性遗传。MFN2基因c.1090C>T位点突变主要临床症状是视神经萎缩等[9]。在本研究的核心家系中,3个患者均无视神经萎缩表现,兄妹都表现出较早的儿童期发病,目前已经采用足部外科矫形逆转其手足畸形,并配合矫形鞋等维持其肌肉的自主能力[10]。
CMT目前无特异性的治疗,大部分患儿诊断明确后仅行物理康复疗法。有学者认为对于CMT病足踝畸形僵硬或复杂者可联合多种术式分阶段矫形治疗,或者联合应用外固定架(如Ilizarov外固定器)逐步矫正畸形[11,12]。Lee等认为神经肌肉病变造成的足踝畸形多存在肌力不平衡,仅仅采用Ilizarov等外固定架治疗虽可矫正畸形,但无法长期维持,所以需行肌力平衡手术(如肌腱延长或移位手术)或关节融合手术以维持长期效果[13]。应用Ilizarov技术治疗多类严重足踝畸形,不仅是畸形矫正满意,而且最大限度地保留了足的外形与功能,同时降低了皮肤和神经血管损伤的风险,减少了并发症,是一项安全可靠的矫治方法[14]。该患者目前通过手术进行肢体畸形的矫正,手术治疗原则是纠正足部畸形,重建和平衡足踝肌力从而减轻痛苦,改正足的位置,避免接触和压疮,手术治疗以获得跖行、无痛行走的足部功能为目的,同时矫正骨性畸形、平衡肌力。
本研究基于患者的临床表型,在一个核心家系中检测并确诊了3例MFN2基因单碱基杂合突变的患者。之前的报道显示此突变主要是以视神经萎缩为主要临床症状,与本研究中报道的四肢畸形不尽相同,也证实了CMT具有明显的临床异质性。CMT尽管目前尚无逆转病情的特异性治疗方法,但外科矫形手术等对症支持治疗可以改善运动功能、提高生活质量、增加患儿自信心促进身心发育,其远期预后仍需长期大样本随访研究。
