17例髓鞘少突胶质细胞糖蛋白抗体相关疾病患者的临床分析
2021-01-07周少旦卜秀群韦馨娴陆婉杏李洵桦胡瑞婷
周少旦,付 桔,卜秀群,韦馨娴,陆婉杏,李洵桦,胡瑞婷
髓鞘少突胶质细胞糖蛋白(Myelin oligodendrocyte glycoprotien,MOG)是少突胶质细胞表面和胞体中的一种糖蛋白,仅在脑、脊髓和视神经等部位中枢神经系统表达,其作用机制尚未明确,但研究已证实MOG抗体在中枢神经自身免疫性疾病中具有重要作用[1]。过去研究认为MOG抗体仅伴随多发性硬化(multiple sclerosis,MS)和视神经脊髓炎(neuromyelitis optica,NMO)等中枢神经系统脱髓鞘疾病出现[2]。随着分子生物学的不断发展,加深了MOG抗体的认识,目前研究认为MOG抗体相关疾病(MOG antibody-associated disease,MOGAD)是一种独立的疾病状态,主要表现为复发或单相病程的视神经炎(optic neuritis,ON)、脊髓炎、脑炎、脑脊髓炎、脑膜脑炎,脊液或(和)血清MOG抗体阳性,可与MS、水通道蛋白4(APQ4)抗体阳性的视神经脊髓炎谱系疾病(neuromyelitis optica spectrum disorder,NMOSD)、急性播散性脑脊髓膜炎(acute disseminated encephalomyelitis,ADEM)等疾病存在部分临床症状重叠,但其发病机制、临床表现、治疗方案和预后不同[3]。本文通过收集中山大学附属第一医院17例MOG抗体阳性MOGAD患者的临床资料并进行回顾性分析,探讨MOGAD患者临床特点及影像学表现。
1 临床资料
通过回顾性方法收集2017年1月-2019年6月就诊于中山大学附属第一医院神经内科17例MOGAD患者临床资料,包括性别、年龄、病史、起病形式、临床症状、MRI表现、神经电生理检查、脑脊液检验、抗核抗体谱、血沉、甲状腺抗体、治疗方案及预后等。MOGAD诊断标准(2020年MOGAD诊断和治疗中国专家共识):(1)临床上表现为ON(包括慢性复发性炎性视神经病变)、脑炎、脑干脑炎、脑膜脑炎、脊髓炎等一种或几种;(2)与CNS脱髓鞘相关的MRI或电生理检查结果;(3)血清或(和)脑脊液MOG抗体阳性[4]。

2 结 果
2.1 一般资料 17例MOGAD患者中,男性11例,女性6例,男女比例为1.83∶1,发病年龄为(27.76±12.22)(10~54)岁。3例有病毒性脑炎/脑膜脑炎病史(3/17,17.65%),1例有地中海贫血病史(1/17,5.88%),1例有肺结核病史(1/17,5.88%),12例无特殊病史。5例患者发病前2 w内有明确前驱感染史(5/17,29.41%),其中4例为急性上呼吸道感染症状,1例为明确的口腔溃疡病史。
2.2 临床特点 10例患者表现视物模糊(8/17)、视野缺损(2/17)、视物变形(2/17)等ON症状(10/17,58.82%),单侧和双侧视神经受累各5例。9例患者表现肢体乏力(9/17)、肢体麻木(8/17)、大小便障碍(3/17)等脊髓受累症状(9/17,52.94%)。10例患者表现头痛、头晕、反应迟钝、记忆力下降、精神行为异常、共济失调、癫痫等脑部受累症状(10/17,58.82%),其中癫痫和头痛各4例,反应迟钝、记忆力下降、精神行为异常2例,共济失调1例。1例表现为典型ADEM症状,主要表现为头痛、呕吐、四肢无力麻木、大小便障碍等症状。
2.3 辅助检查 17例患者均行腰椎穿刺,其中2例脑脊液(cerebrospinal fluid,CSF)压力>180 mmH2O(2/17,11.76%),所检CSF均为无色透明,8例白细胞不同程度增高,波动于(29~480)×106/L(8/17,47.06%)。9例CSF蛋白增高(9/17,52.94%),2例CSF蛋白大于1 g/L(2/17,11.76%),CSF糖均正常,1例CSF氯化物降低(1/17,5.88%),IgG指数波动于0.243~0.724。17例患者血清均检测出MOG抗体(17/17,100%),抗体滴度为(1∶10~1∶320)。其中7例CSF检测出MOG抗体(7/17,41.18%),抗体滴度为(1∶1~1∶32)。1例血清MOG抗体滴度为1∶100,脑脊液MOG抗体滴度为1∶3.2,且合并CSF抗NMDAR抗体(1∶3.2)及GFAP抗体阳性(1∶32)。1例合并CSF寡克隆区带抗体阳性(1/17,5.88%)。1例合并CSF及血清APQ4抗体阳性(1/17,5.88%)。5例血沉增快(5/17,29.41%),7例抗核抗体阳性(7/17,41.18%)。11例患者行甲状腺抗体检测,其中5例为甲状腺抗体阳性(5/11,45.45%)。
2.4 影像学特点 17例患者MRI均发现病灶(17/17,100%),主要表现为颅脑、脊髓、视神经、脑膜等部位T2WI、T2FLAIR序列斑片状高信号影,部分出现边缘强化或斑片状强化。8例患者视神经MRI提示双侧或单侧视神经异常信号,可见视神经局限性增粗水肿(8/17,47.06%),均为视神经前段,节段>1/2视神经,3例出现斑片状强化。16例病灶累及脑部,主要累及大脑皮质下白质(4/17,23.53%)、丘脑(4/17,23.53%)、侧脑室旁白质(2/17,11.76%)、胼胝体(2/17,11.76%)、脑干(中脑1/17、脑桥4/17、延髓5/17)、小脑(1/17,5.88%)、脑膜(2/17,11.76%)。8例病灶累及脊髓,主要累及颈髓(7/17,41.18%)、胸髓(4/17,23.53%)、脊髓圆锥(1/17,5.88%),其中5例表现为>3个节段的长节段脊髓炎,3例表现为<3个节段的短节段脊髓炎(见图1~图3)。
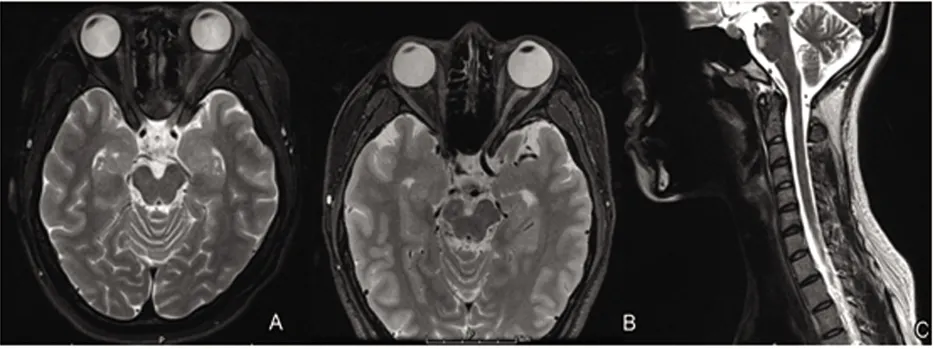
图1 MOGAD患者MRI表现。A:T2WI示双侧视神经眶内段长T2信号影,左侧较右侧增粗肿胀明显;B:T2FLAIR示左侧视神经高信号影,长度>1/2视神经;C:T2WI示脑桥腹侧长T2信号影

图2 MOGAD患者脊髓MRI表现。A:T2WI示颈2~7椎体水平脊髓内可见长T2信号影;B:T2WI示颈2-3椎体水平脊髓内可见长T2信号影

图3 MOGAD头部MRI表现。A、B:T2FLAIR示左侧额顶叶皮质下白质大片高信号影,可见斑片状强化;C:T2FLAIR示右侧大脑脚、脑干高信号影;D:T2FLAIR示胼胝体、扣带回、额叶皮质下白质高信号影
2.5 神经电生理检查 15例患者行视觉诱发电位(VEP),其中8例提示双侧视觉通路异常(8/15,53.33%),3例提示单侧视觉通路异常(3/15,20%),主要表现为P100波潜伏期延长,波幅下降。9例患者行体感诱发电位(SEP),5例提示异常(5/9,55.56%)。
2.6 治疗和预后 17例患者入院均予甲强龙冲击治疗(500~1000 mg/d),13例患者同时予静脉注射免疫球蛋白(intravenous immunoglobulin,IVIG)冲击治疗5 d[0.4 g/(kg·d)],激素逐渐减量,维持3~6个月。16例患者症状在1个月内均有改善,1例患者症状改善不明显,予血浆置换治疗3次后症状改善。电话及临床随访1 y内,6例出现复发(6/17,35.29%),其中1例加用吗替麦考酚酯序贯治疗,1例加用硫唑嘌呤序贯治疗,4例患者再次予激素冲击,症状改善。
3 讨 论
目前MOG在中枢神经系统的作用尚未明确,有研究认为MOG作为细胞黏附因子,通过调节少突胶质细胞微管的稳定性,从而发挥生物学效应[5]。研究证实,MOG抗体在NMDAR抗体阳性的自身免疫性脑炎、病毒感染后CNS自身免疫性炎性疾病、典型MS等疾病中具有重要的作用[6,7]。目前最新研究认为MOGAD是MOG抗体介导的CNS炎性脱髓鞘疾病,是一种新的有特异性治疗策略和方法的疾病状态,其临床特征不同于典型MS、NMOSD、ADEM等其他CNS炎性脱髓鞘疾病,但可存在临床特征交叉重叠[3]。
国外研究发现MOGAD可发生于任何年龄段,儿童多发,成人发病中位年龄为30岁左右,女性多见[8]。也有部分研究发现MOGAD中,男性发病率较高[9],我国学者研究提示男女发病比例为13∶9[10]。本研究男女发病比例为1.83∶1,男性较多见,发病年龄较早,而MS、NMOSD均是女性多发,发病年龄为20~40岁,儿童少见,此结果提示MOGAD发病年龄及性别不同于MS、NMOSD[3,10]。MOGAD常见症状为ON,其次为脊髓炎、ADEM、脑干脑炎、脑膜脑炎等,部分患者可表现为癫痫。MS患者ON主要累及单侧,以短节段视神经损害为主。APQ4抗体阳性ON可累及双侧或单侧视神经,常累及视交叉或视神经后段,节段常长于视神经1/2,症状较重,常以视物模糊为主要症状,易复发。而MOGAD相关性ON主要累及双侧视神经,以前段视神经为主,节段较长,较少累及视交叉,表现为视物模糊、视野缺损、视乳头水肿等,44%~83%患者可出现症状复发,预后较好[4,8,11]。本组患者中10例出现NO症状,累及单侧和双侧视神经各5例,均为视神经前段长节段损害,8例视神经MRI提示双侧或单侧视神经前段局限性增粗水肿,节段>1/2视神经,3例出现斑片状强化。VEP检查提示约73.33%患者出现视神经P100波潜伏期延长,波幅下降,以双侧受累为主,与之前研究结果一致,提示部分患者出现临床症状前已存在视神经损害。与AQP4抗体阳性的NMOSD相比,MOGAD视神经受累症状复发率较高,但其功能预后较好[12]。儿童MOGAD发病年龄较早,主要表现为ADEM或类ADEM症状,极少数表现为极后区综合征,复发间隔时间较长[13]。本组患者中1例38岁男性患者临床症状表现为典型ADEM。
MOGAD患者MR病灶缺乏特异性,主要累及侧脑室旁白质、双侧丘脑、脑桥、脊髓、视神经等部位[12]。MOGAD患者视神经MR表现与APQ4阳性的NMOSD比较,视神经增粗水肿更明显,多位于视交叉前部,节段较长,可出现强化[14]。侧脑室旁白质斑片状病灶不如MS典型,无明显垂直于侧脑室长轴的特征[8]。NMOSD患者脑部病灶多位于极后区或脑干,脑膜强化少见。与NMOSD比较,MOGAD患者大脑灰质及皮质旁白质受累较常见,部分患者可出现脑膜强化[4,14]。研究报道,60%的儿童患者可出现双侧丘脑的病变,也有部分儿童患者出现小脑脚病变[15]。MS患者脊髓病灶主要以短节段受累为主,常位于脊髓偏侧,NMOSD脊髓病灶常为长节段病灶,多位于中央管周围,极少累及脊髓圆锥。而MOGAD患者脊髓炎主要累及颈胸段脊髓,部分累及腰髓和脊髓圆锥,可出现长节段和短节段病灶,以不连续短节段病变更常见,横断面病灶可见于脊髓中央或周围[4,10]。本组17例患者头部及脊髓MRI均发现病灶,主要表现为累及颅脑、脊髓、视神经、脑膜等部位T2WI、T2FLAIR序列斑片状高信号影,可部分出现边缘强化。47.06%的患者出现脊髓受累,以颈胸段为主,长节段脊髓炎和短节段脊髓炎均出现,1例累及脊髓圆锥,提示MOGAD影像学特点不同于MS、NMOSD。
早期研究认为MOG抗体仅存在于血清中,随着研究的不断深入,发现CSF也可检出MOG抗体,但检出率较外周血清低[3]。本组17例患者血清均可检出MOG抗体,滴度为(1∶10~1∶320),而CSF检测出MOG抗体仅有7例,抗体滴度为(1∶1~1∶32),提示血清MOG抗体检测比CSF更为敏感且抗体滴度更高。研究发现MOGAD患者血清MOG抗体滴度与疾病的严重程度、复发率、复发间隔时间存在相关性[8]。本组病例电话随访1 y中,6例患者复发,血清MOG抗体滴度为(1∶32~1∶100),提示抗体滴度可能与疾病复发存在相关可能。MOGAD患者CSF或血清中可检测出其他自身抗体,本组发现1例MOGAD患者合并血清抗NMDAR抗体及GFAP抗体阳性,1例患者合并CSF及血清APQ4抗体阳性,约半数患者合并血清其他自身抗体阳性,提示MOGAD可合并其他自身免疫性疾病。
目前MOGAD尚无明确指南,故MOGAD的诊断标准和治疗方案等尚存在争议。国内外研究的治疗方案均是根据NMOSD、MS等其他中枢神经系统自身免疫性疾病的治疗经验制定,对于是否长期使用激素或免疫抑制剂尚未明确[4]。研究证实,急性期给予糖皮质激素和IVIG冲击治疗,大多数患者预后良好,部分患者遗留后遗症,61.8%患者可出现复发[8]。国外学者发现免疫抑制剂或激素长期维持可减少MOGAD的复发率,但目前缺乏大样本数据[15]。本组17例患者入院时均予激素冲击治疗,13例同时予IVIG冲击治疗,16例症状在1个月内均有改善,1例症状改善不明显,予血浆置换治疗3次后症状改善,与此前国内外研究结果一致。电话随访1 y中,6例出现复发,复发率为35.29%,其中2例加用免疫抑制剂,4例再次予糖皮质激素冲击治疗后症状改善。与既往研究相比,本组患者复发率较低,可能原因为随访时间较短或病例数较少。
综上,MOGAD是一种独立的疾病,其以血清或脑脊液MOG抗体为生物标志物,临床表现多样,与MS、NMOSD、ADEM等其他CNS炎性脱髓鞘疾病存在交叉重叠,大多数患者急性期免疫治疗有效,部分出现复发,复发率可能与血清或脑脊液MOG抗体滴度存在相关性,长期糖皮质激素或免疫抑制剂治疗方案尚存在争议。本研究存在一定局限性,样本量较少,随访时间较短,且缺乏儿童患者,后续研究需扩大样本量,纳入多中心研究,增加随访时间。
