基于密度泛函理论的燃煤飞灰未燃尽碳与烟气砷作用机理
2020-12-30凌杨,吴江
凌 杨, 吴 江
(1.上海电力大学 能源与机械工程学院, 上海 200090;2.上海理工大学 能源与动力工程学院, 上海 200093)
燃煤电厂锅炉烟气成分复杂,其中含有大量空气污染物,若不经处理而直接排放至大气中,则会对人类生产生活环境造成恶劣影响。相比较传统污染物而言,燃煤烟气中的痕量元素污染物,如砷(As)、硒(Se)、汞(Hg)、铅(Pb)等,由于其含量低、难处理、反应机理复杂,越发受到世界各国研究者的广泛关注[1-7]。以砷污染为例,据报道[1,8],燃煤电厂是最大的人为砷污染排放源,有效控制燃煤烟气中各形态砷的排放是全球砷污染治理的关键。
燃煤烟气中砷的形态众多,主要有单质砷As,以及氧化态砷如AsO、AsO2和As2O3等[9-11]。其中,三价态砷(As3+)的氧化物As2O3,热力学性质稳定,不易解离,且具有剧毒,对人体的伤害最大[8,12]。此外,烟气砷会对电厂选择性催化还原(selective catalytic reduction, SCR)脱硝装置造成干扰,引起SCR催化剂砷中毒,进而影响电厂经济效益[13-15]。Lu等[16]选取某电厂连续运行一年的工业级SCR催化剂(其主要成分为V2O5-MoO3/TiO2)进行催化活性实验,催化剂中砷质量分数高达7%,在350 ℃下平均脱硝反应效率仅能保留70%。此外,燃煤烟气脱硫石膏作为电厂副产物,具有诸多工业用途[17-19]。而烟气中的砷污染物若富集于脱硫石膏中,将严重影响产品的二次利用。Liu等[20]对中国20个省份的共70份烟气脱硫石膏样品进行分析,发现脱硫石膏中砷含量普遍在15 mg/kg左右,且以毒性最强的三价砷(As3+)为主,浸出实验也显示了持续砷释放的潜在风险。综上,开发有效技术手段控制烟气中砷污染含量,无论是对生态环境还是对电厂运行经济性而言,均具有重要现实意义。
飞灰作为燃煤电厂的副产物,产量稳定,成本低廉,是一种有潜力的吸附剂[21-23]。若能采用飞灰作为基础吸附剂,对其进行改性处理,并用于烟气污染脱除领域,则可达到废弃物资源化利用之目的。Wang等[5]在某1000 MW电厂SCR系统出口进行飞灰喷射试验,以期对烟气中的痕量元素砷(As)、硒(Se)、铅(Pb)等进行协同脱除。结果显示烟道内喷射改性飞灰可提高电厂静电除尘器(electrostatic precipitator, ESP)和湿法烟气脱硫(wet flue gas desulfurization, WFGD)系统的脱除效率,三种痕量元素污染排放量可降低30%-74%。Li等[24]实验研究了飞灰对气态As2O3的吸附脱除作用。结果表明,在不同电厂收集的飞灰样品均对As2O3有一定的捕获作用,烟气砷的捕获在低温条件下为物理吸附和化学吸附的协同作用,而在高温条件下则以飞灰中有效矿物组分的化学吸附为主。然而,其研究工作主要局限于实验阶段。飞灰中成分复杂,常含有金属/非金属氧化物,如SiO2、Al2O3、Fe2O3、CaO、MgO等[25,26],以及在锅炉炉膛中未参与化学反应而随烟气携带至烟道下游的未燃尽碳(unburned carbon, UBC)[27]。有必要通过理论计算,单独分析飞灰中各组分与烟气砷的作用机制,从而深刻理解气态砷向颗粒态的转化和迁移规律,为指导优化电厂除尘设备运行并提高其脱砷效率提供依据。He等[28,29]构建未燃尽碳模型并进行密度泛函理论(density functional theory, DFT)计算,详细讨论了表面缺陷、表面活性氯基团和酸性气体氛围等对单质汞吸附的影响规律,理论结果与实验现象相一致。Qin等[30]详细讨论了碳质表面含氧官能团的存在对飞灰未燃尽碳吸附单质汞的影响规律,发现含氧官能团可有效提高相邻碳原子活性,且官能团上的碳、氢原子数目对汞吸附特性影响较大。基于密度泛函理论的量子化学计算近年来被广泛运用于污染物脱除研究领域[31-34]。然而,目前,关于飞灰未燃尽碳组分与烟气砷作用机理的理论研究报道还较少。
本研究基于密度泛函理论,构建未燃尽碳理论模型,并对其吸附气态单质砷As及其氧化物AsO、AsO2和As2O3进行了详细讨论,结合吸附能、原子间距和Mulliken布局分析等探究吸附质与载体的作用规律。最后根据态密度和前沿分子轨道分析,以期解释砷氧化物与未燃尽碳的优先吸附结构和作用机理。
1 计算模型与方法
1.1 模型构建
图1所示的锯齿型碳原子团簇结构被证明是模拟碳质表面的有效模型[32,35]。底面和两个侧面添加氢原子使之饱和,将上方碳原子保持裸露以模拟未燃尽碳表面。对构建的初始模型进行几何优化得到最终构型,记为模型A。后续的吸附操作均以模型A为基底。将表面碳原子依次标记为C(1)-C(9),方便讨论。

图1 未燃尽碳模型示意图
1.2 计算方法
本研究的密度泛函理论计算,基于Materials Studio软件中的DMol3模块进行,采取广义梯度近似(generalized gradient approximations, GGA)下的Perdew-Burke-Ernzerhof(PBE)泛函刻画电子相关交换关联作用。SCF(self-consistent field)自洽场计算的能量收敛标准为2.0×10-5Ha;最大力收敛标准0.04 Ha/nm;最大位移变化0.0005 nm。所有电子都被考虑在计算体系内。采取双数值轨道基组加d轨道极化函数为基底。全局轨道截断选取0.4 nm。采取0.005 Ha的拖尾效应(smearing)以加速收敛。在几何优化的模型基础之上,进行能量值自洽计算与Mulliken布局分析,获得原子间距与Mulliken电荷。通过计算吸附能评价体系的稳定性,其定义如下[30]:
Eads=EAB-EA-EB
(1)
式中,Eads为体系的吸附能(eV),EAB为吸附质B吸附于载体A后的总能(eV),EA为载体A的总能(eV),EB为吸附质B的总能(eV)。一般地,吸附能Eads越小(负),则吸附效果越好,吸附构型越稳定[36]。
2 结果与讨论
2.1 单质砷及其氧化物
烟气中气态AsO、AsO2和As2O3分子的建模与几何优化结果如图2所示[12],相应的键长和Mulliken电荷见表1和表2。AsO分子显然呈现直线构型,其中,As-O键长为0.1667 nm。稳定构型的AsO2呈现“V”字型对称结构,其中,As-O键长为0.1680 nm,键角∠O(2)-As-O(1)为127.537°。当As2O3分子呈现空间对称的三角双锥结构时,其总能最低,是热力学稳定的优先构型。其As-O和As-As键长分别为0.1897和0.2452 nm。AsO2分子中的砷化合价最高,失去最多电荷,Mulliken电荷为0.906 e,AsO分子中的砷失去电荷最少,Mulliken电荷为0.493 e,而As2O3介于两者之间。
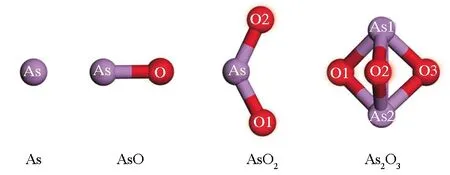
图2 气态单质砷及其氧化物结构示意图
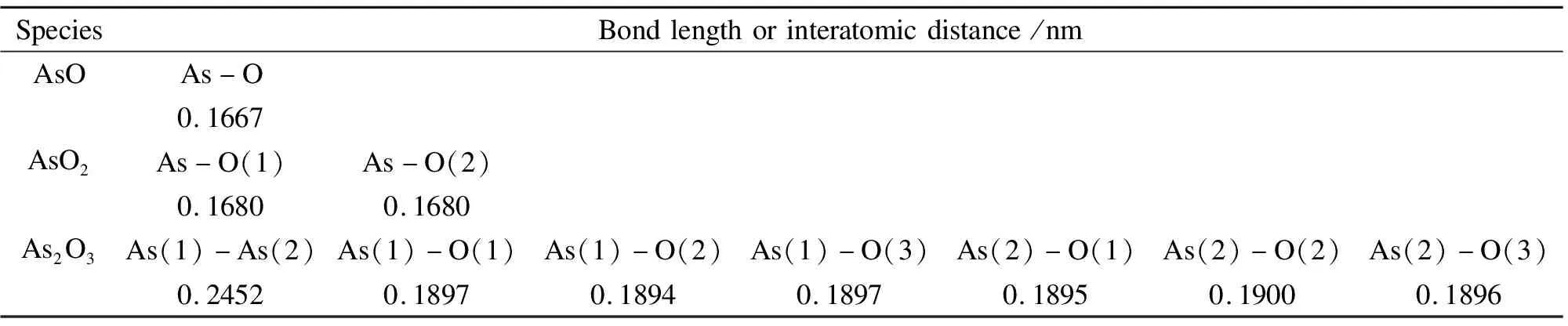
表1 气态砷氧化物分子的键长或原子间距
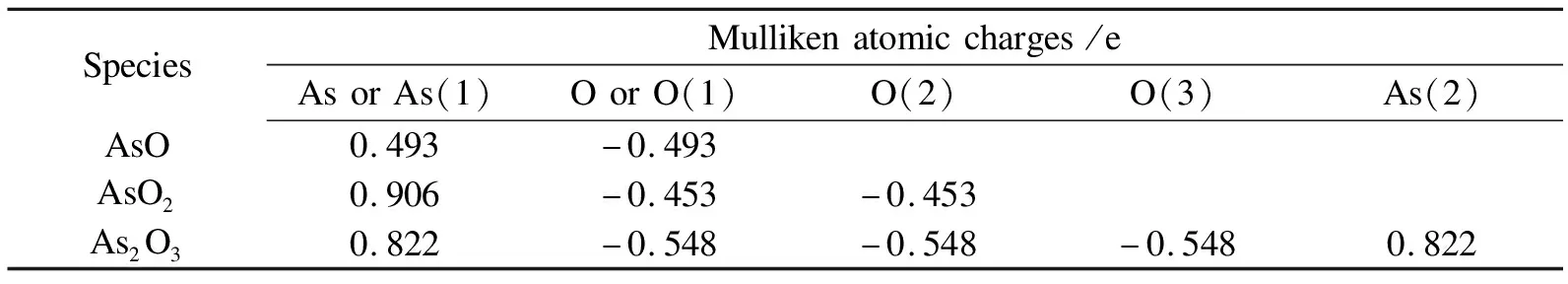
表2 气态砷氧化物分子的Mulliken电荷分布
2.2 单质砷及其氧化物在未燃尽碳表面的吸附
气态单质砷As在未燃尽碳表面吸附的初始构型见图3。考虑到空间对称性,且为避免边缘的碳活性点位对结论的影响[29],选择C(3)桥位、C(4)顶位和C(5)桥位为初始吸附点位,并依次记为I、II和III。将单质砷As分别置于上述吸附点位,并进行几何优化,最终吸附构型及吸附能见图4。初始和最终构型的对应关系见表3,相关键长(原子间距)和Mulliken电荷见表4、表5和表6(下同)。

表3 几何优化结果与相应吸附能

表4 不同吸附构型下未燃尽碳表面键长
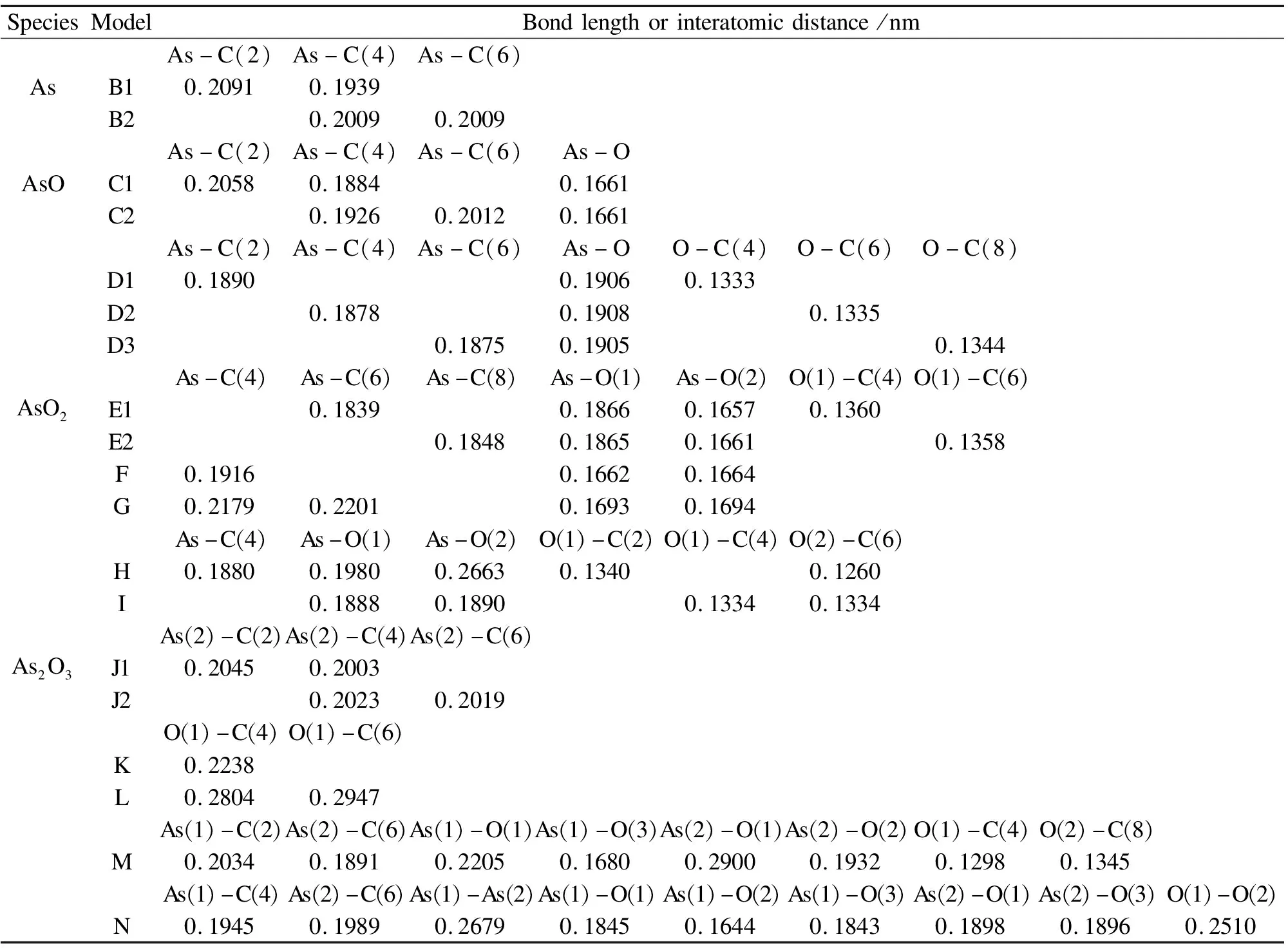
表5 吸附质的键长或原子间距
表6 不同吸附构型的Mulliken电荷分布
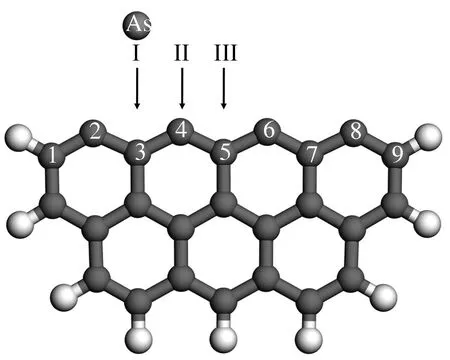
图3 气态单质砷吸附于未燃尽碳的初始构型

图4 气态单质砷吸附于未燃尽碳的最终构型
无论初始构型是碳桥位或碳顶位,单质砷As最终都倾向于吸附在碳质表面的桥位,并与桥位相邻的两个碳原子成键,形成稳固的构型(图4)。模型B1与模型B2吸附能差异不大,为(-5.95)-(-5.88)eV。单质砷吸附于C(3)桥位的吸附能最低,在该构型(模型B1)中,As-C(2)和As-C(4)键长分别为0.2091和0.1939 nm。C(1)-C(2)与C(2)-C(3)键长变化不大,而C(3)-C(4)与C(4)-C(5)键则相比较于洁净碳质表面(模型A,图1)呈现拉长的趋势,由0.1386和0.1384 nm分别变为0.1427和0.1427 nm,表现出单质砷As与相邻碳原子较强的相互作用。从Mulliken布局分析可知,与砷原子直接接触的表层碳原子C(2)与C(4)的电荷分布有较大变化,均获得了较多电荷,分别变为-0.053和-0.095 e。而次表层碳原子C(3)相比较于洁净未燃尽碳表面而言,失去较多电荷,变为0.209 e。单质砷由孤立态下的电中性,变为带正电荷(0.132 e)。这说明了表面碳原子与单质砷的吸附作用,有部分的电荷从单质砷转移到了碳质表面。通过比较模型B1、B2可知,模型B2中的砷原子失去的电荷相对较少,其Mulliken电荷为0.115 e,这也和两者的吸附能结论一致。
图5为AsO分子在未燃尽碳表面的吸附初始构型。考虑三种初始位置:(a)AsO垂直吸附,且以砷原子首先与碳质表面接触;(b)AsO垂直吸附,且以氧原子首先与碳质表面接触;(c)AsO平行吸附于碳质表面。对于垂直吸附的情况,依然采用 C(3)桥位、C(4)顶位和C(5)桥位作为初始吸附点位,初始构型记为I、II、III、IV、V和VI。而对于AsO平行吸附的情况,则以砷原子依次置于C(3)、C(4)、C(5)和C(6)原子上方为初始构型,标记为VII、VIII、IX和X。最终优化结果与相应吸附能见图6。

图5 气态AsO分子吸附于未燃尽碳的初始构型
如图6所示,当AsO分子以垂直形式吸附,且以砷原子首先接触未燃尽碳表面,最终将形成亚稳定构型,体系吸附能较高,在(-4.17)-(-4.10)eV,其值也略高于单质砷的吸附。与单质砷吸附情况类似,此时的AsO分子依靠砷原子吸附在碳桥位C(3)或C(5)上,并与桥位相邻的碳原子成键。在砷原子外靠近真空的一侧悬挂有一个氧原子,构成了几乎垂直的吸附构型。以吸附能较低的模型C1为例,As-O键为0.1661 nm,相较于孤立态AsO而言,键长基本保持不变,碳质表面与氧原子作用较弱。As-C(2)和As-C(4)键长分别为0.2058和0.1884 nm,其相比较于单质砷吸附于C(3)桥位时的相应键长,均有所缩短。当AsO分子吸附在C(3)桥位时,C(2)与C(4)原子均获得一定负电荷,达到-0.023和-0.086 e。而C(3)原子则失去较多负电荷,变得带正电,为0.187 e。该现象与未燃尽吸附单质砷的情形类似,但程度上略有下降,这也与两者吸附能差异相一致。值得注意的是,当气态AsO分子处于以氧原子先接触未燃尽碳表面的垂直初始构型,或是以平行接触的初始构型时,其经过几何优化后的最终构型,均表现为图6中的模型D结构。在该结构中,砷、氧原子分别与相邻的碳原子结合形成化学键,构成稳定的吸附结构,吸附能较低,在(-7.87)-(-7.64)eV。该吸附能值均明显低于模型C中的数值,说明模型D是气态AsO分子吸附于未燃尽碳表面的优先结构。以吸附能最低的模型D2为例,As-C(4)与O-C(6)键分别为0.1878和0.1335 nm。而As-O键为0.1908 nm,相比较于AsO孤立态分子中的0.1667 nm而言被显著拉长,可归因于未燃尽碳表面不饱和碳原子与砷、氧原子更强的相互作用。AsO与碳质表面结合,将优先形成一种特殊的五元环结构,如模型D2中的As-O-C(6)-C(5)-C(4)五元环,该结构更加稳定,相比较于其他吸附构型有明显优势。这一规律在下述AsO2或As2O3分子吸附的情形中也有体现。
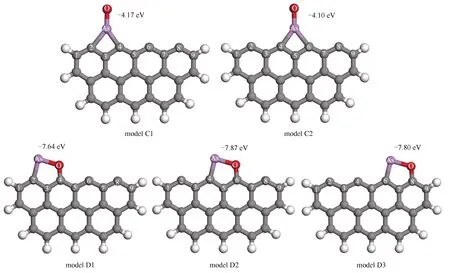
图6 气态AsO分子吸附于未燃尽碳的最终构型
考察气态AsO2分子在未燃尽碳表面的初始吸附结构,具体见图7。

图7 气态AsO2分子吸附于未燃尽碳的初始构型
类似地,选取三种初始吸附位置:(a)AsO2以垂直的形式与碳质表面接触,将O(1)原子分别位于C(4)、C(5)和C(6)原子上方时,记为初始构型I、II和III;(b)AsO2分子以“V”字型与未燃尽碳表面接触,将砷原子分别位于C(4)和C(5)原子上方,记为构型IV和V;(c)AsO2分子以倒“V”字型与未燃尽碳表面接触,将砷原子分别位于C(4)和C(5)原子上方时,记为构型VI和VII。最终的几何优化结果与相应吸附能见图8。
如图8所示,当AsO2分子以垂直形式接触未燃尽碳表面时,最终均呈现模型E的结构,吸附能在(-6.36)-(-6.08)eV。以吸附能较低的模型E1为例,首先氧原子O(1)与相邻碳原子C(4)结合,O(1)-C(4)键长0.1360 nm,接着砷原子将表面碳C(6)结合成键,As-C(6)键长0.1839 nm。另外一个氧原子O(2)悬挂于砷原子上,不与碳表面成键,As-O(2)键长0.1657 nm。通过观察可以发现,模型E中吸附质与未燃尽碳接触的局部结构和模型D类似,吸附能也明显低于单质砷的吸附,表现出较好的吸附特性。当AsO2以“V”字型吸附,且以砷原子首先接触未燃尽碳表面时,得到模型F和模型G,结构稳定性较差,吸附能分别为-3.98和-3.24 eV,明显高于单质砷吸附的情形。可见气态AsO2分子以砷原子首先接触未燃尽碳表面,并不是最佳吸附路径。而当AsO2呈现倒“V”字型以两个氧原子首先接触未燃尽碳表面时,表现出优异的吸附性能(模型H和模型I)。在模型H中,AsO2分子在未燃尽碳表面发生解离现象,形成一个AsO和表面活性氧的独特构造,吸附能低至-10.65 eV,说明该结构是AsO2在未燃尽碳表面吸附的最佳构型。在该构型中,由AsO2解离形成的AsO与未燃尽碳表面形成较强化学键作用,As-C(4)和O(1)-C(2)键长分别0.1880和0.1340 nm,这也和模型D或模型E中与未燃尽碳表面成键的砷、氧原子规律类似,预示着砷、氧原子与表面临近碳原子形成特殊五元环的稳定结构,是砷氧化物与碳质表面作用的优先形式。气态AsO2分子在未燃尽碳表面的次优先构型如模型I所示,此时两个氧原子分别于表面碳成键,而砷原子只与两个氧原子成键而不再和表面碳接触。此时,吸附能达-7.25 eV,虽高于最优构型(模型H),但仍优于模型E的情形。其值也可与AsO分子的最佳吸附构型(模型D)相当。通过对比可以发现,当单质砷及其氧化物中的氧原子参与吸附成键时(无论砷原子与表面碳成键与否),形成的吸附能均较低,构型要稳定于砷原子单独与未燃尽碳表面成键的情况。
在各形态烟气砷中,三价态砷(As3+)的毒性最大,是燃煤电厂烟气砷污染控制的关键。讨论热力学结构稳定的三角双锥As2O3分子构型在未燃尽碳表面的吸附,如图9所示,选取三种初始吸附结构:(a)As2O3以As(1)-As(2)垂直于未燃尽碳表面的形式进行接触,将As(2)原子分别位于C(4)和C(5)原子上方时记为初始构型I和II;(b)As2O3以As(1)-As(2)平行于碳表面,且以单个氧原子O(1)先接触未燃尽碳,将氧原子O(1)分别位于C(4)和C(5)原子上方时记为初始构型III和IV;(c)As2O3同样以As(1)-As(2)平行于碳质表面,但由两个氧原子O(2)和O(3)先接触未燃尽碳,将氧原子O(2)分别位于C(4)和C(5)原子上方,记为初始构型V和VI。对上述初始结构依次进行几何优化,其最终结果与相应的吸附能见图10。

图9 气态As2O3分子吸附于未燃尽碳的初始构型
当气态As2O3分子以As(1)-As(2)垂直于未燃尽碳表面,以As(2)先接触未燃尽碳表面时,所得最终吸附结构如图10中的模型J所示。此时As2O3分子的结构保持完整,吸附能较高,在(-3.60)-(-3.59)eV,明显高于模型B和模型C的情形,且与模型F和模型G相当。以模型J1为例,As(2)原子吸附于C(3)桥位,与相邻碳原子C(2)、C(4)成键,As(2)-C(2)和As(2)-C(4)键长分别为0.2045和0.2003 nm,相较于氧原子参与吸附的情形(模型D、模型E和模型H),有显著伸长。当砷原子单独与未燃尽碳表面成键时,吸附能较高,而有氧参与成键时,则吸附构型将更加稳定。当As2O3以As(1)-As(2)平行于碳表面,且以单个氧原子O(1)先接触未燃尽碳时,所得最终结构为模型K和模型L。此时As2O3仍保持结构完整,吸附效果较差。模型K吸附能仅-0.83 eV,As2O3分子位于C(4)顶位,O(1)-C(4)间距0.2238 nm。此时O(1)的Mulliken电荷-0.538 e,与孤立态As2O3中的差异不大,预示着该构型中吸附质与未燃尽碳之间较弱的相互作用。在模型L中,As2O3分子位于C(5)桥位,吸附能虽相较于模型K有所降低,但仍仅有-3.59 eV的水平。然而,当气态As2O3分子以两个氧原子O(2)和O(3)先接触未燃尽碳时,所得结构(模型M和模型N)变化较大。在模型M中,As2O3分子在未燃尽碳表面呈现较完全的解离状态,产生了一个AsO2分子与一个AsO分子,并分别与表面碳结合。由As2O3裂解所产生的小分子与未燃尽碳表面形成较强的相互作用,吸附能低达-10.64 eV,与模型H相当。类似地,模型M中形成两个彼此独立的五元环结构,即As(1)-O(1)-C(4)-C(3)-C(2)环与As(2)-O(2)-C(8)-C(7)-C(6)环,该稳定结构的形成是体系吸附能较低的主要原因。As2O3也有可能形成诸如模型N所示的次稳定结构,两个砷原子As(1)和As(2)分别与碳原子C(4)和C(6)成键,键长分别为0.1945和0.1989 nm,吸附能-5.37 eV,明显高于模型M的情形,且也高于AsO、AsO2分子的最佳吸附构型(模型D和模型H)。
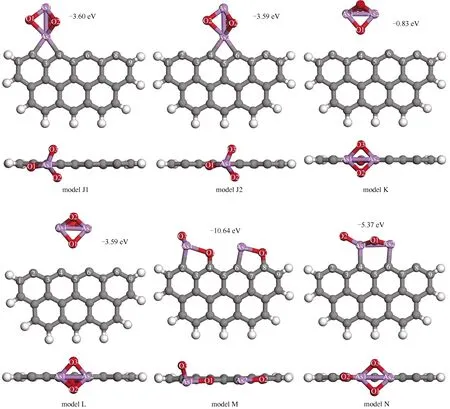
图10 气态As2O3分子吸附于未燃尽碳的最终构型
2.3 电子结构
经上述讨论,AsO或AsO2分子吸附于未燃尽碳表面时,均优先形成一个局部的五元环结构。而对于三价态砷(As3+)而言,热力学稳定的三角双锥As2O3,若不发生解离,则吸附能较高,与未燃尽碳的相互作用较弱。而当As2O3吸附于未燃尽碳表面后,若能解离成小分子,如AsO和AsO2等,则吸附能将显著降低,系统更加稳定,此时体系仍优先形成局部的五元环结构,可见该结构的重要性。以AsO作为吸附质,选取具有代表性的模型D2进行态密度(density of states, DOS)分析,并以洁净碳质表面模型A作为参考,其结果见图11和图12。
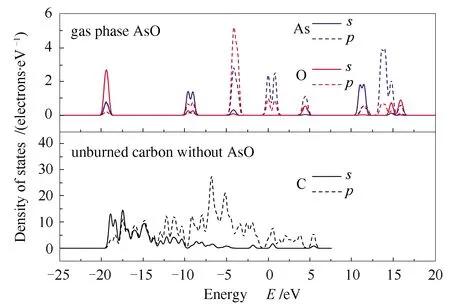
图11 气态AsO分子与洁净未燃尽碳的态密度
图11分别为孤立的气态AsO分子,以及未经吸附的洁净碳质表面(模型A)的态密度分布。从图11中可以看出,孤立AsO分子于费米能级以下的电子态能量分布,在位于-19、-9和-4 eV处呈现尖锐峰值,局域化程度较高,说明AsO分子中的砷原子与氧原子表现出较强的共价作用。其中,位于-19 eV左右的尖峰,以O 2s电子轨道为主,伴随有As 4s态的贡献。位于-4 eV左右强烈的尖峰,则由O 2p和As 4p轨道电子云重叠引起。而未经吸附的洁净未燃尽碳,其电子态分布在(-20)-0 eV能量范围内均呈现非局域特性,此为碳的sp杂化能级典型特征。
图12为AsO分子吸附于未燃尽碳表面,并形成模型D2时,体系中As、O和C元素的态密度分析。从图12中可以看出,当吸附完成时,各元素的态密度能量相较于未吸附时而言,均有所降低。吸附态的AsO和未燃尽碳载体,均在-22 eV处形成局域的尖峰,表明该处成键,主要是C 2s和O 2s的强烈作用。吸附态AsO在(-15)-0 eV能量条件下,电子态非局域特性增强,说明孤立态AsO中的强烈共价键作用被未燃尽碳削弱,这也与吸附态AsO中分子键被拉长的结论相一致。仔细观察吸附后的未燃尽碳,在(-7)-(-2)eV,与吸附态AsO的态密度呈现良好对应,表现出较强的共价作用,可归因于C 2p、O 2p和As 4p电子云重叠。
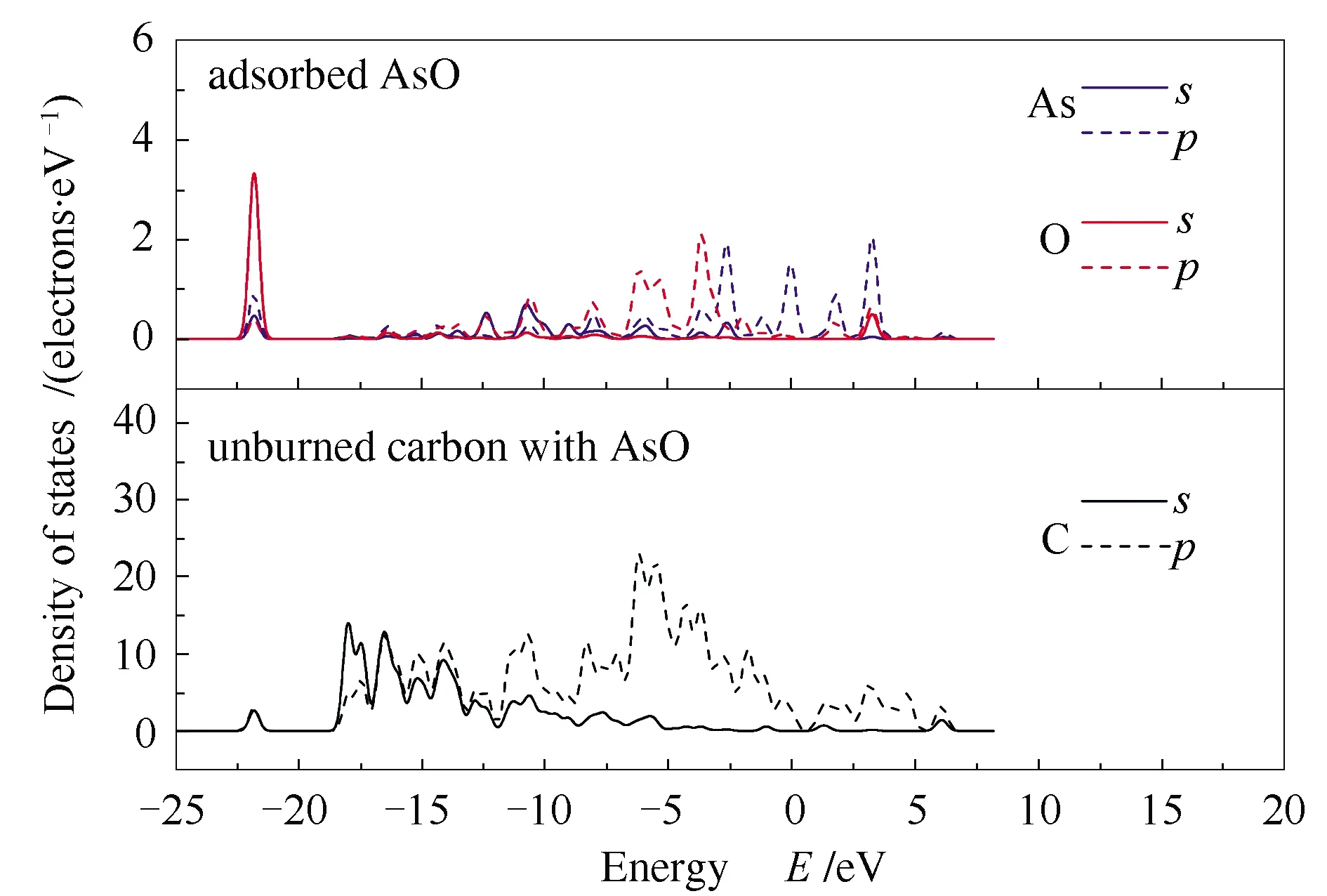
图12 吸附态AsO与吸附后未燃尽碳的态密度
基于前沿分子轨道理论(frontier molecular orbital theory)对未燃尽碳吸附AsO分子的过程进行了分析,其结果见图13。洁净未燃尽碳(模型A),其最低未占分子轨道(lowest unoccupied molecular orbital, LUMO)与最高占据分子轨道(highest occupied molecular orbital, HOMO)分布类似,均主要由边缘的碳原子,即C(2)、C(4)、C(6)和C(8)提供,说明边缘碳原子具有较高的化学反应活性,是吸附发生的优良场所[29]。气态AsO分子的HOMO中,砷、氧原子形成p轨道电子云重叠,形成共价作用。而LUMO则主要由砷原子提供,表明该位置是良好的电子接受体。当AsO分子吸附于未燃尽碳并形成模型D2后,从体系的HOMO可以看出砷原子与C(4)周围形成较强的电子云重叠,砷原子从未燃尽碳获得较多的电荷而被还原,这也与Mulliken布局分析一致(0.493→0.345 e)。与氧原子直接接触的C(6)原子被显著氧化,失去大量电荷,从未吸附前的-0.046 e变为吸附发生后的0.326 e。未燃尽碳与砷、氧原子强烈的相互作用,在局部产生形如As-O-C(6)-C(5)-C(4)的独特五元环稳定结构,是导致了体系吸附能降低的主要原因。
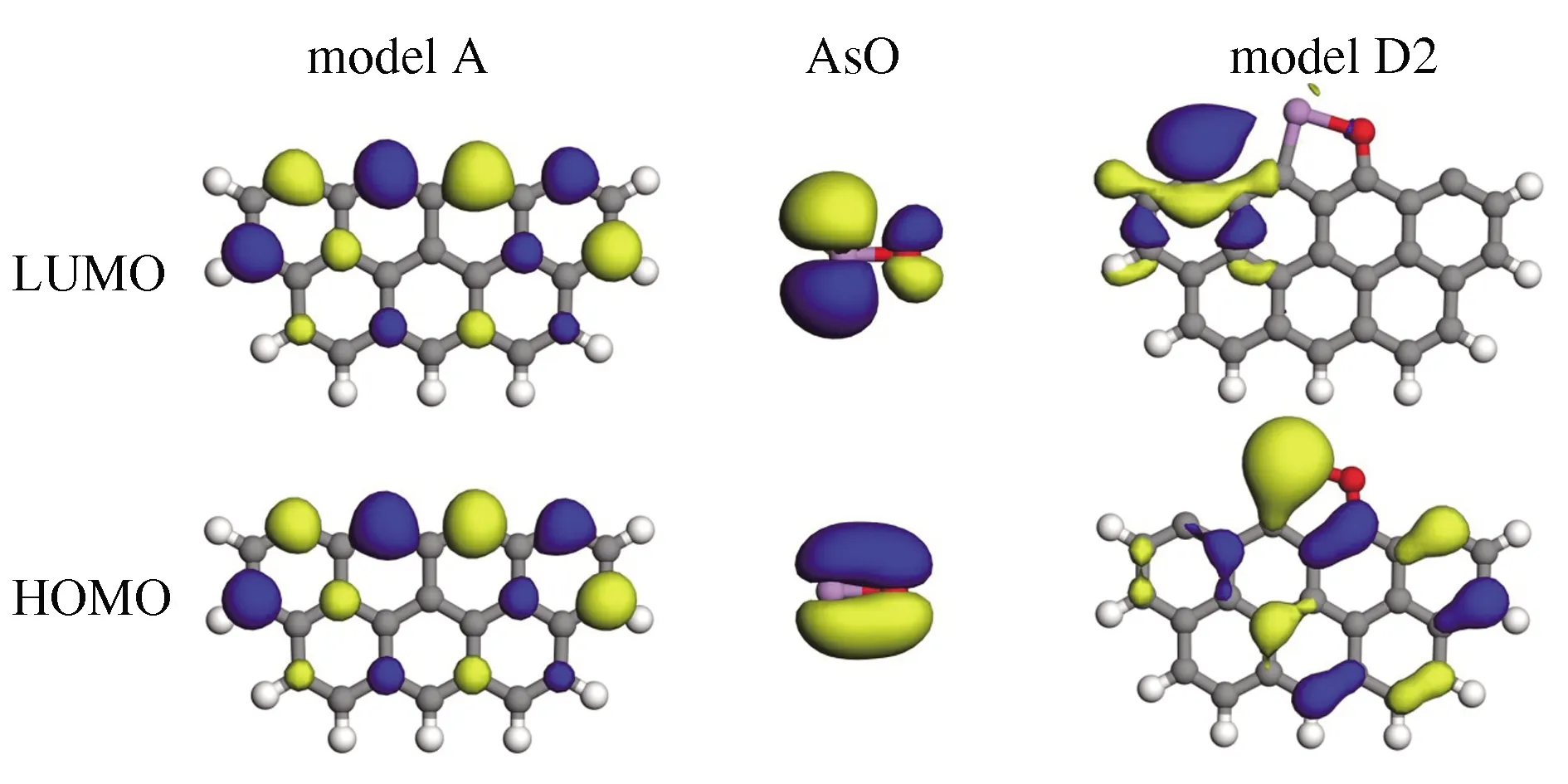
图13 气态AsO分子吸附于未燃尽碳的前沿分子轨道
3 结 论
飞灰中未燃尽碳成分对单质砷As及其氧化物AsO、AsO2和As2O3均具有热力学稳定的吸附功效。单质砷优先吸附于碳桥位,吸附能在(-5.95)-(-5.88)eV。当AsO分子中的砷、氧原子分别与碳原子成键时,吸附构型最稳定,吸附能在(-7.87)-(-7.64)eV。当AsO2在未燃尽碳表面解离,形成一个AsO和表面活性氧并分别与表面碳原子成键时,结构最稳定,吸附能低达-10.65 eV。当As2O3分子在未燃尽碳表面不发生解离时,吸附能较高,甚至高于单质砷的情形。毒性最强的三价态砷(As3+),相比较于As、AsO和AsO2而言,不易发生吸附,是燃煤电厂烟气中砷污染物脱除的关键。然而,当热力学结构稳定的三角双锥As2O3分子以两个氧原子首先碰撞未燃尽碳表面时,将优先形成AsO和AsO2小分子,分别与表面碳吸附并成键,此时体系吸附能相较于未解离的情形而言,明显降低,达到-10.64 eV。未燃尽碳与AsO或AsO2小分子的结合较紧密,一对成键的砷、氧原子倾向于和表面碳形成局部的五元环结构。因此,将热力学结构稳定的三角双锥As2O3分子,催化裂解为AsO、AsO2小分子,将是烟气砷污染控制的有效技术途径。
猜你喜欢
杂志排行

燃料化学学报的其它文章
- Study on the environmental effects of heavy metals in coal gangue and coal combustion by ReCiPe2016 for life cycle impact assessment
- 复合聚并协同脱除燃煤颗粒物及颗粒态重金属的中试研究
- In-situ reaction between arsenic/selenium and minerals in fly ash at high temperature during blended coal combustion
- 典型钙/镁基吸附剂对二氧化硒吸附特性研究
- Speciation analysis of arsenic in coal and its combustion by-products in coal-fired power plants
- 燃煤烟气中As、Se、Pb的形态分布及S、Cl元素对其形态分布的影响