单中心回顾性吉兰巴雷综合征诊治分析
2020-12-12尚贤金丁小牛
尚贤金,徐 昕,李 刚,丁小牛
(皖南医学院第一附属医院 弋矶山医院 1.神经内科;2.微机中心,安徽 芜湖 241001)
吉兰巴雷综合征(Guillain-Barré syndrome,GBS)早在1916年就已报道,临床表现为四肢远端肌力减退,肌腱反射减弱或消失的自身免疫性周围神经病,同时出现脑脊液蛋白-细胞分离现象。随着近些年GBS新的疾病亚型的认识,如咽-颈-臂无力、下肢截瘫型、双侧面瘫伴感觉异常类型以及重叠类型等,学者们开始认识到GBS是累及周围神经系统的一类自身免疫性谱系疾病[1-2]。因此,新的GBS疾病诊断与分类标准更强调依据患者临床特征早期诊断,方法简单更全面,但目前国内临床应用较少,缺乏相关疾病亚型诊断数据[3-5]。本研究参考新的GBS诊断标准,利用弋矶山医院微机中心临床诊治样本库,分析和总结皖江地区GBS临床特点和各亚型分型数据。
1 资料与方法
1.1 一般资料 通过回顾性检索弋矶山医院HIS系统2014年1月~2019年5月出院诊断GBS或格林巴利综合征的神经内科住院患者,排除不符合或其他可解释病因病例(见图1)。患者的临床数据仅用于回顾性病例分析研究,患者身份信息均已匿名化,符合皖南医学院弋矶山医院伦理要求。
1.2 推荐诊断标准 入组GBS患者分型包括四肢无力型、急性截瘫型、咽-颈-臂无力型、面瘫伴肢体远端感觉障碍型,以及Miller-Fisher综合征或Bickerstaff脑干脑炎,具体标准参考文献[2]。
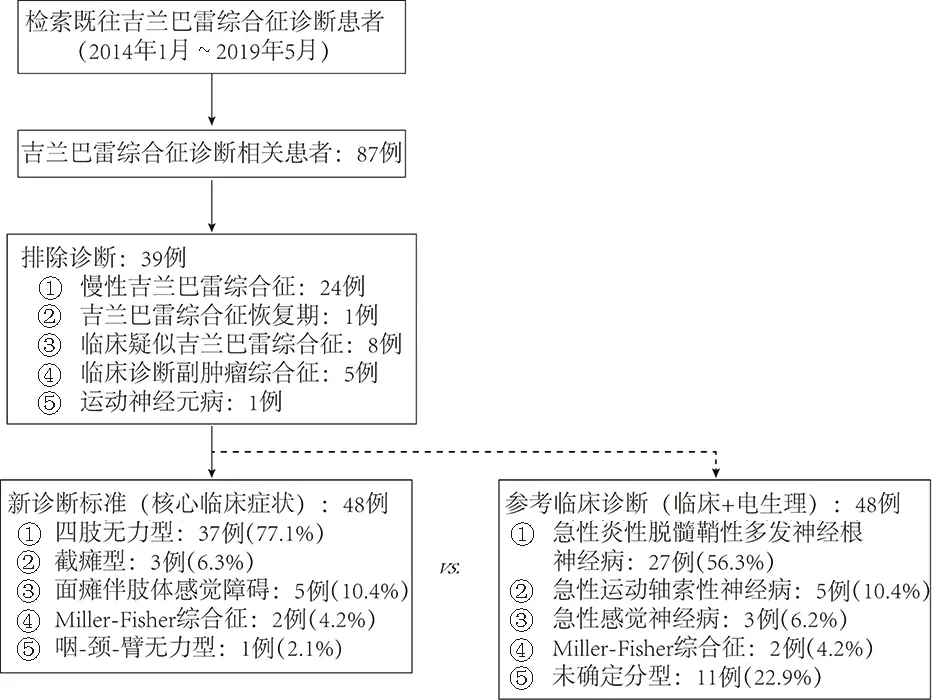
图1 GBS患者诊断流程图
1.3 收集数据和临床结局 收集患者人口学资料、临床特征、脑脊液检查、血液化验,神经电生理检查、详细药物治疗过程,以及6个月左右随访临床结局。随访临床结局采用通用GBS残疾量表评定,方式是通过电话随访或门诊住院复诊情况判断,量表分为6个水平:0分代表健康;1分代表有轻微症状,但是可以跑动;2分代表无辅助下可以步行10米距离,但是不能跑动;3分代表可在辅助下步行10米,并通过空旷区域;4分代表卧床、转移需要轮椅辅助;5分代表日常需要人工辅助通气才能维持身体需要;6分代表死亡。将结局恢复好转定义为6个月左右GBS残疾量表评分≤2分。
2 结果
共检索87例急性GBS患者,排除慢性GBS(包括急性发病的慢性GBS)患者(24/87,27.6%),另有患者(8/87,9.2%)临床资料不全被剔除,同时排除副肿瘤综合征(5/87,5.7%),GBS恢复期和运动神经元病患者各1例(2/87,2.3%)。最终纳入的48例患者的GBS分型和临床特征数据见图1、表1。

表1 GBS谱系疾病患者临床特征(n=48)
GBS患者发病前呼吸道感染(20/48,41.7%)和/或胃肠道感染(8/48,16.7%)常见,47.9%(23/48)患者可能合并其他感染或不明诱因。患者因双下肢症状起病者多见(18/48,37.5%),29.2%(14/48)出现上、下肢运动和感觉同时受累,部分患者突出表现肢体或其他部位疼痛(18/48,37.5%),神经检体发现有4例患者(8.3%)肢体肌腱反射正常,未发现肌腱反射升高患者。脑脊液检测发现有8例(16.7%)患者正常,但多集中在症状出现后1周内(中位数6天);神经传导速度检测发现34例(70.8%)患者有周围神经损害证据,6例(12.5%)无明显异常,其余8例(16.7%)患者因为病情原因未行检查,但综合患者临床表现和脑脊液蛋白-细胞分离现象,排除其他可解释疾病后临床仍考虑GBS,相关诊断分类如图1所示。
有75.0%(36/48)患者同意使用丙种球蛋白治疗,相对于未使用丙种球蛋白治疗患者(12/48,25%),其临床治疗好转(GBS残疾量表评分≤2分),差异无统计学意义(86.1%vs. 100%;χ2=0.670,P=0.413),2例死亡患者分别因呼吸肌及咽喉肌无力继发肺部感染死亡。
3 讨论
本研究回顾分析我院神经内科既往诊治病例,采用GBS谱系疾病诊断与分类标准,综合患者临床发病特点,总结了皖江地区GBS各类型发病比例和疾病诊治特点。
研究发现GBS患者成年男性居多,超过半数患者存在感染诱因,临床症状和体征复杂多样,符合既往研究报道,所以针对不典型症状、体征的周围神经疾病患者,临床病因需要仔细鉴别[1]。尽管脑脊液和神经电生理检查能够协助诊断,但研究发现GBS患者发病后1周内,仅约80%患者腰穿脑脊液有典型的蛋白-细胞分离现象;神经传导检查异常高峰期通常发生在肢体无力出现后2周内,早期电生理检查可能正常[6,7]。本研究发现16.7%GBS患者脑脊液检查蛋白浓度正常,检查中位时间为6天;12.5%患者神经传导速度正常,检查过早可能是主要原因。因此,对于考虑GBS诊断的周围神经病患者,需要警惕临床检查结果假阴性现象,必要时进行动态腰穿和电生理检测协助诊断和治疗。
目前临床广泛应用的GBS诊断标准主要参考1990年修订标准,该标准纳入GBS必要和强烈支持诊断的临床证据,以及需要鉴别的其他疾病,用于临床研究目的[8]。2001版修订的GBS诊断共识将疾病分为感觉运动型、纯运动神经、Miller-Fisher综合征和脑干类型[9]。上述分类不包括近年发现的一些GBS不典型或症状重叠患者。2011年布雷顿诊断标准强调依赖神经电生理、腰穿辅助检查,临床诊断特异度提高的同时,降低了诊断灵敏度[10]。因此,2014年新发表GBS诊断分类和标准全面地描述了四肢无力型GBS,Miller-Fisher综合征及其亚型,包括GBS各谱系疾病的诊断特点,依据患者临床特征综合诊断,提高了诊断的灵敏度[2]。基于新诊断标准,尽管部分患者没有或早期缺乏典型的神经电生理表现,仍能准确分型。本研究发现四肢乏力或腱反射减弱或消失的典型症状起病患者发病率(77.1%)高于其他类型,是最常见的发病类型,截瘫型、面瘫伴肢体感觉障碍型、Miller-Fisher综合征亚型和咽-颈-臂无力型分别达到了6.3%、10.4%、4.2%和2.1%,区别于美国地区各亚型的2%、1%、5%和3%[2],中国台湾地区存在7% Miller-Fisher综合征、7%脑干类型、5%咽-颈-臂型和5%多颅神经炎型[11],还有荷兰研究报道6%截瘫型和1%咽-颈-臂无力型[10]。不同地区人群发病率差异原因考虑以下两点:一是环境气候和地区感染病原体差异,因为分子模拟机制是GBS发病的主要机制之一,不同神经部位受累可导致不同的临床症状;二是不同地区人群或个体的遗传易感性差异所致[12-13]。其具体原因仍需进一步大样本、国际多中心研究阐明。
新诊断标准依据患者核心临床表现进行诊断,不仅提高GBS早期诊断率,也有助于更早地进行有效治疗(静脉使用免疫球蛋白等)。然而,以前的诊断标准需要依靠患者临床特点以及脑脊液、电生理检查确定诊断,因此患者诊断资料不全或辅助检查不及时,可能导致个体诊断分型困难。即使依靠电生理结果,也可能发现24%~38%GBS患者亚型诊断有误[14-15]。值得强调的是,新的GBS诊断分类标准将神经电生理和血清抗体检测作为疾病的支持证据,如Miller-Fisher综合征或亚型患者,抗GQ1b或GD1b抗体检测阳性对于其诊断有重要辅助价值[2,16]。本研究中多数患者未进行抗神经节苷脂抗体检测是局限性之一,但由于抗体检查费用昂贵和周期长,在目前GBS要求尽快诊断、及早治疗的情况下,抗体检测临床应用价值有限。另外,单中心回顾性观察研究也是该研究局限性之一。
综上所述,急性炎症性脱髓鞘性周围神经病仍是皖江地区人群最常见的GBS发病类型,同时临床工作中不应漏诊其他亚型,及时诊断和有效治疗仍是患者良好功能预后的前提。
