烟酰胺碳核苷类似物的模块化合成
2020-11-09奚亦凡马莹莹李宏瑞程志浩张永强
奚亦凡, 马莹莹, 李宏瑞, 程志浩, 杨 凯, 张永强
(华东理工大学 药学院,上海 200237)
烟酰胺核苷(Chart 1),通过转化为烟酰胺腺嘌呤二核苷酸(NAD),广泛参与体内氧化还原代谢反应,在细胞物质代谢和能量合成等多种生理活动中发挥着重要作用[1]。烟酰胺核苷的碳核苷类似物,如Tiazofurin和Benzamide Riboside(Chart 1),具有与其相似的三维立体特征和更好的代谢稳定性,可选择性干扰肿瘤细胞异常的物质代谢和能量合成,表现出广谱的抗肿瘤活性[2-9]。例如,Tiazofurin可有效抑制次黄嘌呤脱氢酶(IMPDH),干扰鸟苷酸合成,作为治疗慢性髓系白血病的候选药物,目前处于临床II期研究阶段。
烟酰胺碳核苷类似物,作为抗肿瘤候选药物,结构上通常包含D-(脱氧)核糖和芳(杂)甲酰胺两部分,二者以Csp2-Csp3键(碳核苷键)相连,通常为β构型,具有多手性中心和多亲水性基团的特征。该类结构的合成方法主要有两种,一种是从头合成法,一种是片段偶联法。从头合成法,以芳(杂)环或(脱氧)核糖基的从头构建为特征。如Tiazofurin的合成,需要通过硫化氢与氰基加成、硫代甲酰胺与溴代丙酮酸乙酯的环化等3步反应,从头构建噻唑环(Scheme 1A)[10],也有经氰基与半胱氨酸乙酯环化和进一步芳构化,从头构建噻唑环的报道[11- 12]。Benzamide Desoxyriboside的合成,也曾使用从头合成脱氧核糖基的方法,由于涉及到多手性中心的构建,整个路线需要11步反应[13]。显然,这类方法立足于目标结构,不具有普适性,且步骤相对繁杂,合成效率不甚理想,难以有效应用于多样性烟酰胺碳核苷类似物的快速构建。片段偶联法,以(脱氧)核糖基与糖苷配基的直接偶联为特征,代表着更为直接有效的合成方式,尤其在Benzamide Riboside的合成中,得到了广泛的应用(Scheme 1B)[5-7,14-15]。然而该方法的关键片段偶联步骤,往往使用危险易燃芳(杂)基有机金属试剂与核糖内酯等的亲核加成反应,存在反应条件苛刻、反应官能团兼容性差等缺陷。由此可见,缺乏温和高效通用的模块化合成方法,不利于多样性烟酰胺碳核苷类似物的快速构建,成为制约相关抗肿瘤新药开发的瓶颈。

Chart 1
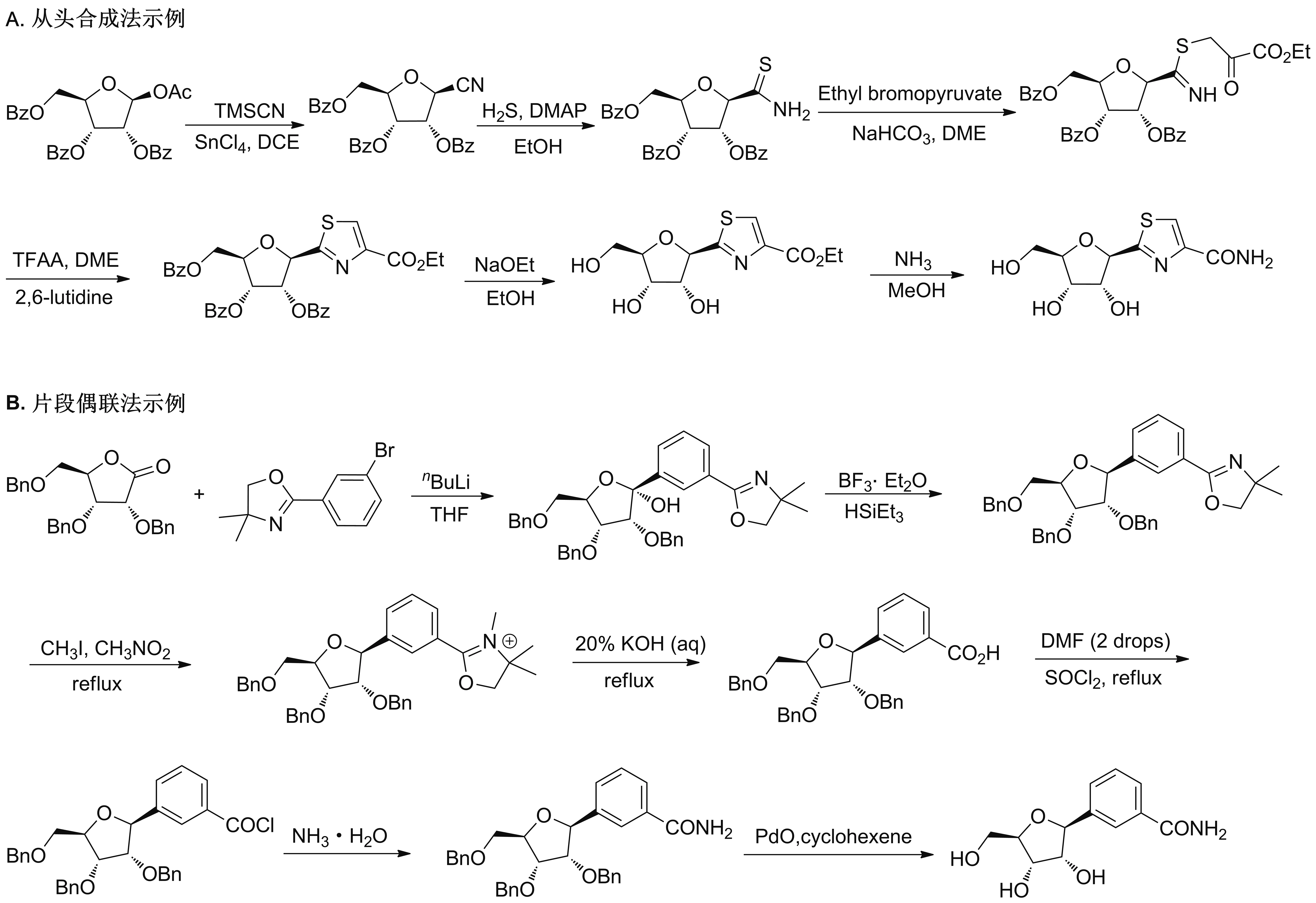
Scheme 1
可见光/金属镍双重催化脱羧偶联反应,通过可见光催化的脱羧反应促进烷基自由基生成,降低了金属镍催化偶联反应的能垒,使其在绿色温和条件下,有效介导Csp2—Csp3键的构建,在药物合成中展现出广阔的应用前景[16-17]。我们团队在该领域也开展了一系列卓有成效的工作,实现了(脱氧)核糖异头羧酸与溴代芳(杂)环的有效偶联[18]。有鉴于此,以核糖异头羧酸合成为起始,利用该反应进行核糖基和糖苷配基的片段偶联,与氰基水解相结合,本研究发展了一种高效实用的苄基保护烟酰胺碳核苷类似物合成新方法(Scheme 2)。该方法,简洁高效,立体选择性优异(β/α>99/1);所用关键砌块溴代芳(杂)甲腈,结构多样,商业易得,化学性质稳定,有助于实现模块化合成。基于该方法,本文初步实现了多样性苄基保护烟酰胺碳核苷类似物6a~6i的合成,其结构经1H NMR,13C NMR和HR-MS(ESI)表征。

Scheme 2
1 实验部分
1.1 仪器与试剂
Autopol I 型旋光仪;ZF-TA 型手提式紫外分析仪;Bruker DPX-400/Avance-500 MHz型核磁共振仪(CDCl3为溶剂,TMS为内标);Finigann MAT8401型高分辨质谱。
4CzIPN(CAS: 1416881-52-1),毕得医药;其余所用试剂均为分析纯或化学纯。
1.2 合成
(1)3的合成[19]
将化合物18.2 g(17.7 mmol)溶解于乙腈(35 mL)中,冷却至-48 ℃,缓慢加入TMSCN 3.3 mL(26.6 mmol)和BF3·Et2O 2.7 mL(21.2 mmol),搅拌下反应15 min(TLC检测)。用饱和NH4Cl溶液淬灭反应,用乙酸乙酯(3×20 mL)萃取,合并有机相,用饱和食盐水(2×20 mL)洗涤,无水Na2SO4干燥,浓缩,残余物经硅胶柱层析(洗脱剂:PE/EA=3/1,V/V)纯化得化合物α-21.9 g,收率25%和β-23.8 g,收率50%。
将β-23.8 g(8.8 mmol)溶解在60 mL二氧六环和6 mL浓盐酸中,加热至80 ℃,搅拌下反应4 h(TLC检测)。冷却至室温,将混合物倒入水(20 mL)中,用乙酸乙酯(3×30 mL)萃取,合并有机相,依次用饱和食盐水(2×30 mL)洗涤,无水Na2SO4干燥,浓缩,残余物经硅胶柱层析(洗脱剂:PE/EA/AcOH=100/100/1,V/V/V)纯化得产物33.8 g,收率95%,β/α>99/1。
将α-21.9 g(4.4 mmol)溶解于20%KOH的甲醇(50 mL)溶液中,加热至80 ℃,搅拌下反应4 h(TLC检测)。冷却至室温,浓缩,以6M HCl酸化,用乙酸乙酯(3×50 mL)萃取,合并有机相,依次用饱和食盐水(2×30 mL)洗涤,无水Na2SO4干燥,浓缩,残余物经硅胶柱层析(PE/EA/AcOH=100/100/1,V/V/V)纯化得31.4 g,收率70%,β/α=5/1。
β-3:1H NMR(400 MHz, CDCl3)δ: 7.42~7.19(m, 15H), 4.79(d,J=12.0 Hz, 1H), 4.67(s, 1H), 4.60(d,J=3.1 Hz, 1H), 4.57(d,J=3.0 Hz, 1H), 4.49~4.44(m, 2H), 4.32(dd,J=9.4 Hz, 2.5 Hz, 1H), 4.21(dd,J=11.7 Hz, 1H), 4.14(d,J=4.5 Hz, 1H), 3.97(dd,J=9.3 Hz, 4.5 Hz, 1H), 3.87(dd,J=10.4 Hz, 2.9 Hz, 1H), 3.54(d,J=10.4, 1H);α-3:1H NMR(400 MHz, CDCl3)δ: 7.50~7.10(m, 15H), 4.76(d,J=11.1 Hz, 1H), 4.64(d,J=11.3 Hz, 1H), 4.55(d,J=4.6 Hz, 1H), 4.53(d,J=4.7 Hz, 1H), 4.46(s, 1H), 4.44(d,J=11.7 Hz, 1H), 4.31(dd,J=8.4 Hz, 3.0 Hz, 1H), 4.20(d,J=11.7 Hz, 1H), 4.14(d,J=4.7 Hz, 1H), 3.96(dd,J=8.9 Hz, 4.7 Hz, 1H), 3.82(dd,J=10.0 Hz, 2.9 Hz, 1H), 3.52(d,J=10.0, 1H)。
(2)6a~6i的合成通法
在25 mL干燥的Schlenk反应管中,加入NiBr2(0.02 mmol), bpy(0.024 mmol), K2CO3(0.4 mmol), 4-CzIPN(0.01 mmol),溴代芳(杂)甲腈(0.4 mmol)和3(0.2 mmol)。封闭反应管并通入氮气,注入DMF 10 mL,搅拌下,通氮气15 min。将反应管置于34W蓝色LED条带下照射,搅拌下反应24 h(控温30 ℃左右,TLC检测)。加入30 mL水,用乙酸乙酯(3×20 mL)萃取,合并有机相,依次用饱和食盐水(2×20 mL)洗涤,无水硫酸钠干燥,浓缩,残余物经硅胶柱层析(洗脱剂:PE/EA=5/1,V/V)纯化得中间体5a~5i。
将5a~5i0.1 mmol溶于DMSO(3 mL)中,冷却至0 ℃,加入K2CO3(0.4 mmol)和30%H2O21.8 mL(0.5 mmol),缓慢升至室温,搅拌下反应1 h(TLC检测)。冷却至0 ℃,用饱和Na2S2O3溶液淬灭反应,用乙酸乙酯(3×20 mL)萃取,合并有机相,依次用饱和食盐水(2×20 mL)洗涤,无水Na2SO4干燥,浓缩,残余物经硅胶柱层析(洗脱剂:PE/EA=1/1,V/V)纯化得6a~6i。
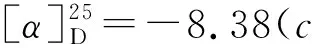
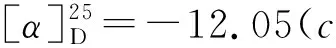
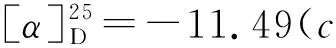
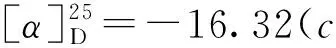
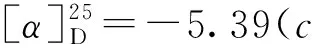

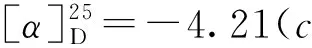
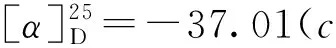
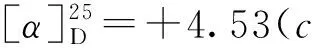
2 结果与讨论
2.1 氰基水解与核糖异头羧酸合成
如Scheme 2所示,在三氟化硼乙醚作用下,3,4,5-三-O-苄基-1-乙酰氧基-D-核糖可原位生成氧鎓离子,与TMSCN发生亲核加成,仅需15 min即可完全转化为中间体2,且α和β两种异构体易于分离。因此,在下一步氰基水解反应中,直接以α-2和β-2为底物,对反应条件进行了研究(表1)。该步转化通常有酸和碱两种反应条件。在酸性条件下,β-2易于水解,以95%收率,立体保持的转化为核糖异头羧酸3(β/α>99/1);而α-2,却主要生成核糖异头甲酰胺3′,且异头位发生部分构型翻转(β/α=4/1)。这可能是因为,在酸性条件下水解时,α-2可通过形成氢键,生成热力学稳定的六元环中间体I和II,所以第一步水解可以很快完成,而第二步水解却难以有效进行;而强酸性条件下烯醇(胺)中间体III的生成,导致了异头位构型翻转,生成了热力学更为稳定的β-3′(Scheme 3)。然而,在碱性条件下,α-2却可以有效发生两步水解,同时伴随着异头位构型翻转(β/α=5/1)。我们推测,异头位去质子化和烯醇(胺)负离子中间体VI的生成,导致该位点构型翻转,生成了热力学更为稳定的β构型中间体VII;而该中间体的生成,是第二步水解易于进行的主要原因(Scheme 4)。

表1 3,4,5-三-O-苄基-1-氰基-D-核糖水解反应研究

Scheme 3

Scheme 4

表2 光镍双重催化脱羧偶联反应研究
2.2 光镍双重催化脱羧偶联反应与碳核苷键构建
在前期研究中,课题组对核糖异头羧酸和溴代芳(杂)环的光镍双重催化脱羧偶联反应,已进行了系统的研究,获得了优化反应条件[18]。在本研究中,以Benzamide Riboside的合成为模板,重点对溴代苯底物进行了筛选,以确保苯甲酰胺糖苷配基可有效生成。首先尝试了间溴苯甲酰胺,反应不能有效进行,这可能和酰胺可以与镍配位,干扰原有反应进行有关(表2)。
表3 化合物5g的水解反应研究
2.3 氰基水解与芳(杂)甲酰胺糖苷配基的构建
缺电子含氮芳杂甲腈的氰基,电正性更高,容易发生二次水解,生成羧酸;此外,氮原子的潜在配位效应,客观上弱化了多种过渡金属催化氰基水解反应与这类结构的兼容性。因此,相关碳核苷中间体5e~5i的水解更具挑战性。有鉴于此,我们以化合物5g的水解为模板,对反应条件进行了简单筛选。过渡金属催化的方法[20-22],包括PdCl2/CH3CONH2, H2O/THF和CuI/Cs2CO3/DBU/CH3NO2/H2O/100 ℃,的确效果不佳。而无过渡金属参与的水解方法效果相对较好,其中过氧化氢促进的碱性水解方法[K2CO3/H2O2(30%)/DMSO/rt][23],条件温和,效果良好,对立体选择性没有影响,成为发展烟酰胺碳核苷类似物模块化合成方法的优先选择。
从商业易得的3,4,5-三-O-苄基-1-乙酰氧基-D-核糖出发,经氰基取代,氰基水解为羧基,可见光催化/金属镍双重催化脱羧偶联反应,氰基水解为酰胺基等4步反应,开发了一项高效通用的苄基保护烟酰胺碳核苷类似物合成新技术,实现了一系列该类化合物的高立体选择性模块化合成。更多苄基保护烟酰胺碳核苷类似物的合成,以及经脱保护反应构建相关化合物库的研究正在进行之中。
