整合宏组学方法研究番茄与玉米秸秆共堆肥生境中的关键微生物及其功能
2020-10-31李俊良朱倩倩张小梅
朱 屹,李俊良,焦 博,朱倩倩,张小梅
(青岛农业大学资源与环境学院,山东 青岛 266109)
0 引言
【研究意义】联合国粮食和农业组织(Food and Agriculture Organization of the United Nations, FAO)统计结果表明,2016 年中国的番茄(Solanum lycopersicum L.)产量超过5 600 万t。与高产作物同时产生的还有近1.3 亿t 的番茄残株,这些有机废物亟需妥善处理[1]。堆肥是处理农业废弃物的有效手段之一。然而,传统堆肥发酵工艺持续时间长、效率低,蔬菜秸秆较农作物秸秆具有的高木质素含量更是加剧了这一问题[2]。同时,由于堆肥发酵是一个微生物生态学过程,物料来源及组成的不同,如蔬菜秸秆较粮食作物秸秆具有更高的含水率、更低的C/N 且富含氮磷钾等植物必需营养元素,将使得其内直接或间接作用的微生物种类差异较大[3]。因此,针对蔬菜秸秆开展具体的堆肥化研究,并有效探明影响腐解的关键微生物及其功能,对进一步优化其发酵工艺具有重要作用[4]。【前人研究进展】前人对秸秆类废弃物堆肥化过程中的微生物群落结构变化研究,多集中在利用保守标记基因(如16S rRNA 等)指纹图谱为基础的分子生物学手段。如贾洋洋等[5]利用限制性片段长度多态性(RFLP)技术研究了玉米秸秆堆肥中细菌的群落结构变化;Liu 等[6]利用变性梯度凝胶电泳(DGGE)技术检测了番茄秸秆堆肥过程中,不同调理剂(生物炭、泥炭沼、沸石等)的添加对细菌群落多样性及其组成的影响。高通量焦磷酸测序技术的产生,为定性、定量地表征生态系统中全部的微生物群落信息提供了可能。该方法灵敏度较高,已被广泛应用于各种复杂生境研究中[7-8]。然而,高通量测序产生的海量数据信息无法表征微生物活性。近年来,新兴的宏蛋白质组学(Metaproteomics)技术则可以原位(in situ)分析微生物的催化活性[9],而整合了焦磷酸测序和宏蛋白质组学的整合宏组学方法(Integrated meta-omics)则将微生物的群落结构及其功能联系了起来[10]。【本研究切入点】目前,整合宏组学方法已成功应用于多种生态系统研究中[8],但尚未见将其应用于蔬菜废弃物处理的研究报道。【拟解决的关键问题】本研究首次将整合宏组学方法应用于番茄秸秆堆肥中,用以揭示堆肥发酵期关键微生物的群落结构、主要的降解酶类及关键微生物的功能,为针对番茄秸秆微生物菌剂的有效复配提供科学指导。
1 材料与方法
1.1 物料来源
所用新鲜番茄秸秆取自青岛农业大学现代农业示范园周边农场;用于调节堆肥参数的辅料——玉米秸秆直接来源于农业示范园内。物料的基本性质如表1 所示。
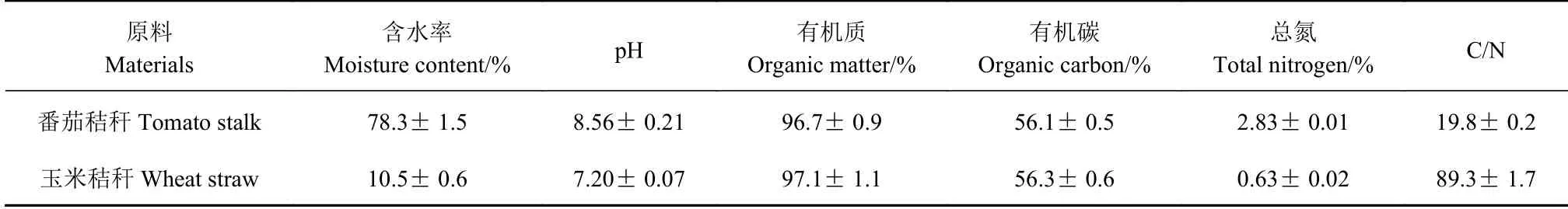
表1 堆肥原料的理化性质Table 1 Physiochemical properties of raw materials for composting
1.2 堆肥试验
堆肥试验于2017 年6~7 月在山东省青岛胶州市青岛农业大学现代农业示范园区(120.1°E,36.4°N)内进行。先将番茄秸秆和玉米秸秆切成2~5 cm 的小段,然后按番茄秸秆和玉米秸秆有机碳为56%,总氮分别为2.8%和0.6%计算,二者按3∶1(m∶m)的比例混合,混合物料C/N 约为25∶1,调节物料含水率为50%~65%,堆成长×宽×高约为5.5 m×1.5 m×1.5 m 的条垛,进行条垛式堆肥研究。
1.3 取样方法
堆肥试验共持续5 周。在发酵堆建堆开始后的第1、2、3、4、5 周(即第7、14、21、28、35 d)分别于堆体20~50 cm 深处、采用“五点取样法”采集样品,样品混匀后分成两部分。一部分自然风干用于理化性质的测定,一部分立刻-20 ℃冻存,用于提取样品总DNA 和酶液的制备、酶学性质分析。
1.4 理化性质测定
为监测发酵堆温度的变化,将5 个1 m 长的温度计从堆体不同的方向和位置分别插入20~50 cm 深,每天同一时刻(14:00)记录堆体内部温度,其平均值作为发酵堆每日温度;同时记录每日环境温度的变化。物料含水率的测定根据所取样品(10 g)在105℃下烘干过夜减少的重量计算得出。样品pH值通过将样品与去离子水按1∶9(W / V)配制,在240 r·min-1下 机 械 振 动1 h 并 静 置30 min 后,用pH 计(Ultra Basic-7,US)测定其上清液所得。利用外加热重铬酸钾法测定样品的有机质含量[11],有机质含量除以换算系数1.724 得到有机碳量[12-13]。样品的总氮量由凯氏定氮法测定[14]。
1.5 DNA 提取和焦磷酸测序
按照PowerSoil DNA Kit 使用说明提取堆肥样品的基因组DNA,每个样品重复3 次。DNA 浓度用Nanodrop(Oxfordshire,UK)在260 nm 波长下测定。质检后的DNA 作为序列扩增用模板,分别用条码融合探针338F(5'-ACTCCTACGGGAGGCAGCAG-3')和806R(5'-GGACTACHVGGGTWTCTAAT-3')覆盖V3~V4 高变区扩增16S rDNA 基因片段[15];用1737F(5'-GGAAGTAAAAGTCGTAACAAGG-3')和2043R(5'-GCTGCGTTCTTCATCGATGC-3')扩增内转录间隔区(Internal transcribed spacer, ITS)rDNA 基因片段[16]。16S rDNA 和ITS rDNA 基因扩增体系和条件按文献[17]进行。将扩增产物统一成等摩尔浓度,用 ABI GeneAmp 9 700(Applied Biosystems, Foster City,CA,USA)和罗氏GS FLX 平台(Roche Life Sciences,Indianapolis,IN,USA)测试分析。通过QIIME(http://qiime.org/scripts/assign_taxonomy.html)对所得序列信息进行质控。
1.6 系统发育分类
质控后的序列信息利用UPARSE 方法进行读长聚类,定义序列相似度≥97%的序列为一个操作分类单元(Operational taxonomic unit, OTU)。16S rDNA 和ITS rDNA 序列比对的参考数据库分别为Silva(http://www.arb-silva.de)和ITS rRNA(http://unite.ut.ee/index.php)。
1.7 酶活测定及统计分析
粗酶液按文献[18]所述方法制备。蛋白酶活性采用Folin-酚法[19];内切纤维素酶和木聚糖酶活性采用二硝基水杨酸(DNS)法测定[20]。所有试验均重复3 次。以相对酶活性(同一实验条件下,各吸光度与最大吸光度的比值)表征各酶活性随时间的变化。酶活性与主要微生物属(平均相对丰度>1%)间的Pearson 相关系数通过SPSS v20.0(SAS Inc., USA)计算,并设定P<0.05 和P<0.01 为显著相关和极显著相关。
1.8 蛋白提取和宏蛋白质组学分析
取第3 周的堆肥样品用于宏蛋白质组学分析。将25 g 堆肥样品、100 mL 去离子水混匀后,置于4 ℃浸提24 h,4 ℃ 10 000 r·min-1离心10 min,收集上清液,并通过0.22 μm 滤膜过滤。滤液再分别通过3 kDa的超滤管(Sigma-Aldrich, St.Louis, MO, USA)超滤浓缩、三氯乙酸沉淀、双蒸水重悬成适当浓度后,按张丽丽等[7]的方法进行胰酶消化和脱盐。获得的肽溶解于10 μL 50%(V / V)乙腈和0.1%(V / V)三氟乙酸溶液中,并通过LTQ-Orbitrap-MS/MS(Thermo Fisher, Pittsburg.PA,USA)进行质谱鉴定。样品酶解后的肽段通过2000 V 的电压喷入质谱仪,转移毛细管的温度为275 ℃。所得数据通过Xcalibur 2.2.0(Thermo Fisher Scientific)进行分析。
1.9 质谱数据的生物信息学分析
从Uniprot 数据库(http://www.uniprot.org)中下载第3 周堆肥样品中丰度较高的几个优势细菌属和优势真菌属(Thermomyces)的蛋白质组数据信息,形成质谱数据比对的参考数据库。用Proteome Discovered software 1.4(Thermo Fisher Scientific)对数据进行比对分析。错配阈值设为0.05。设定当某蛋白在第3 周堆肥样品的3 个重复中被检测到两次及以上时,认为该蛋白存在,3 个平行样的光谱数(Spectrum count)平均值作为该蛋白的平均相对丰度[21]。
2 结果与分析
2.1 堆肥表观变化
为监测发酵堆的表观变化,堆体在第一次取样时进行了纵剖。发现表层(0~20 cm)样本干燥且相对完整;中间层(20~50 cm)有白色菌丝明显可见;而底层(>50 cm)白色菌丝消失。这种分层现象在之前的文献中也有报道[8],表明在没有翻堆的情况下,中间20~50 cm 深度范围内最适合微生物生长[22]。因此,采集该层样品深入分析。
2.2 温度和酶活性的变化
堆温在第1 d 时就迅速由28 ℃升高到55 ℃,且在整个监测期内温度都高达50 ℃以上(图1-a),说明高温堆肥试验成功。监测了与碳氮代谢相关主要酶活性的变化,发现蛋白酶在第7 d 时活性较高,之后下降,至第3 周时酶活性最高,之后又快速下降;而作为植物生物质降解的主要酶类,内切纤维素酶和木聚糖酶在开始的前3 周内酶活性不断上升,之后下降(图1-b)。这与堆肥初期先降解可溶性有机物,堆温快速上升;然后再对较难降解的纤维素和半纤维素类物质降解的规律是一致的[23]。由于第3 周时碳氮代谢主要酶的活性最高,故对该周样品进行了后续高通量焦磷酸测序和Orbitrap 宏蛋白质组学分析。
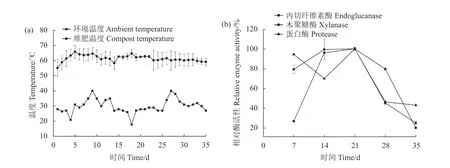
图1 堆肥温度(a)和主要酶活力(b)的变化Fig.1 Changes on temperature (a) and activities of primary enzymes (b) in compost
2.3 微生物群落组成
对第3 周3 个重复样品进行高通量测序,质控后分别获得26 898、30 863 和28 457 条16S rDNA 序列和59 845、61 299、60 128 条ITS rDNA 序列;这些序列分别对应679、703 和681 个细菌OTU 及259、276 和297 个真菌OTU。细菌OTU 被归为6 个主要的门(平均相对丰度>1%)。丰度最高的前4 个门分别为变形菌门Proteobacteria(37.2%)、厚壁菌门Firmicutes(26.4%)、放线菌门Actinobacteria(23.0%)和拟杆菌门Bacteroidetes(8.25%)(图2-a),这与之前的文献报道基本一致[24]。其中,放线菌门下主要的属为嗜热裂孢菌属Thermobifida(16.5%)和糖单孢菌属Saccharomonospora(1.36%);变形菌门下主要的属为海源菌属Idiomarina(15.6%);厚壁菌门的代表属有Limnochordaceae norank(5.40%)、芽孢杆菌属Bacillus(4.25%)和嗜热杆菌属Thermobacillus(1.91%);黄杆菌属Flavobacterium(2.37%)是拟杆菌门的典型代表(图2-c)。相对于细菌群落而言,真菌的群落结构要简单得多。子囊菌门Ascomycota占绝对统治地位,比例为81.3%(图2-b),其中嗜热真菌属Thermomyces 为优势真菌属(相对丰度70.5%)(图2-b)。
对照整个发酵过程中主要(平均相对丰度>1%)微生物的群落结构变化(图3),可以看出,海源菌属Idiomarina 是第1 周时的主要细菌属(相对丰度11.6%),第2 周时基本检测不到,至第3 周时相对丰度又提高至15.6%。第2 周时主要细菌属为Limnochordaceae norank(17.9%)和氢化菌属Hydrogenispora(14.9%)。嗜热裂孢菌属Thermobifida 和嗜热真菌属Thermomyces在发酵开始后丰度逐渐提高。嗜热裂孢菌属Thermobifida第1 周时仅为1.4%,至第2 周时已升至8.9%;嗜热真菌属Thermomyces 也由开始时的10.0%提高至61.1%。至第3 周时,二者丰度分别升至16.5%和70.5%,成为该时期的主导细菌和真菌属。之后二者丰度逐渐降低,至发酵结束时,相对丰度仅为1%左右。
2.4 宏蛋白质组学分析
从图1-b 可以看出,第3 周时植物生物质降解相关的酶(内切纤维素酶和木聚糖酶)活性最高,秸秆降解的主要功能微生物代谢最为旺盛,对该时期样品进行质谱鉴定更能快速定位秸秆降解的关键微生物[7-8]。利用LTQ-Orbitrap-MS/MS 对第3 周的堆肥样品进行分析,所得数据与主要的微生物属(图2-c)蛋白质组进行比对,发现嗜热裂孢菌属Thermobifida是堆肥中的主要功能微生物。堆肥样品中鉴定到的纤维素酶全部由嗜热裂孢菌属Thermobifida 产生,同时还鉴定到源于该属的一种木聚糖酶(P74912)、一种果胶裂解酶(Q47MW8)和两种具有纤维素结合能力的蛋白(Q47PB9 和Q47QG3)(表2)。以上结果说明,嗜热裂孢菌属Thermobifida 在纤维素的结合与降解、半纤维素的降解和果胶裂解过程中发挥重要作用,是堆肥有机质形成的主要参与者,这与之前的文献报道是一致的[7]。
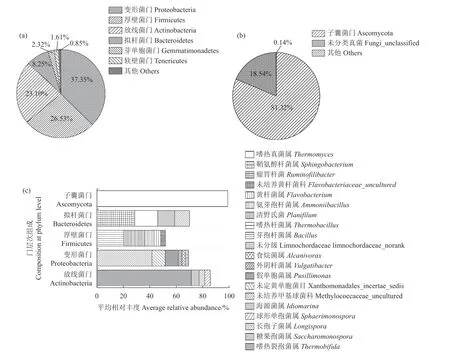
图2 第三周堆肥样品中细菌门(a)、真菌门(b)及主要属(平均相对丰度>1%,c)组成Fig.2 Microbes by bacterial phylum (a), fungal phylum (b) and genera with average relative abundance greater than 1% (c) in 3-week compost sample
鉴定到的木聚糖酶产生者,除嗜热裂孢菌属Thermobifida 外, 还有放线菌门的糖单孢菌属Saccharomonospora;厚壁菌门的清野氏菌Planifilum及子囊菌门的嗜热真菌属Thermomyces,三者所产木聚糖酶的家族和登录号分别为GH10 家族的I1C XX6、GH35 家 族 的A0A1I2PGX8 和GH11 家 族 的O43097(表2)。清野氏菌Planifilum 隶属于高温放线菌科Thermoactinomycetaceae,革兰氏阳性好氧菌,该属菌株最适生长温度35~65℃[25],在蘑菇渣堆肥、干草堆、土壤和水体中广泛分布[26-27]。嗜热真菌属Thermomyces 相对丰度与木聚糖酶活性显著正相关(表3),同时,Thermomyces lanuginosus 被证明因能产生多种耐热半纤维素酶而在多种物料的堆肥发酵过程中大量出现[7]。以上结果说明,嗜热裂孢菌属Thermobifida、糖单孢菌属Saccharomonospora、清野氏菌Planifilum 和嗜热真菌属Thermomyces 是主要的半纤维素降解者。
鉴定到参与蛋白质降解的功能菌主要为糖单孢菌属Saccharomonospora 和海源菌属Idiomarina。糖单孢菌属Saccharomonospora 最早分离自蘑菇渣堆肥中,其胞外蛋白酶多耐热(如最适温度60 ℃)且偏碱性(如最适pH 8.0)[28]。在本研究结果中,源于Saccharomonospora 的蛋白酶主要有两类:丝氨酸蛋白酶(C7MXV7、A0A1V9AC37)和胰蛋白酶(C7MV18、C7MTG2)。这些都是典型的碱性蛋白酶。除此之外,还鉴定到一种源于海源菌属Idiomarina的丝氨酸内切蛋白酶(A0A094IT45)。该蛋白酶的鉴定、Idiomarina 的相对丰度与蛋白酶活性间的显著正相关性结果(表3)表明,海源菌属Idiomarina 在堆肥发酵蛋白质的降解过程中发挥重要作用。
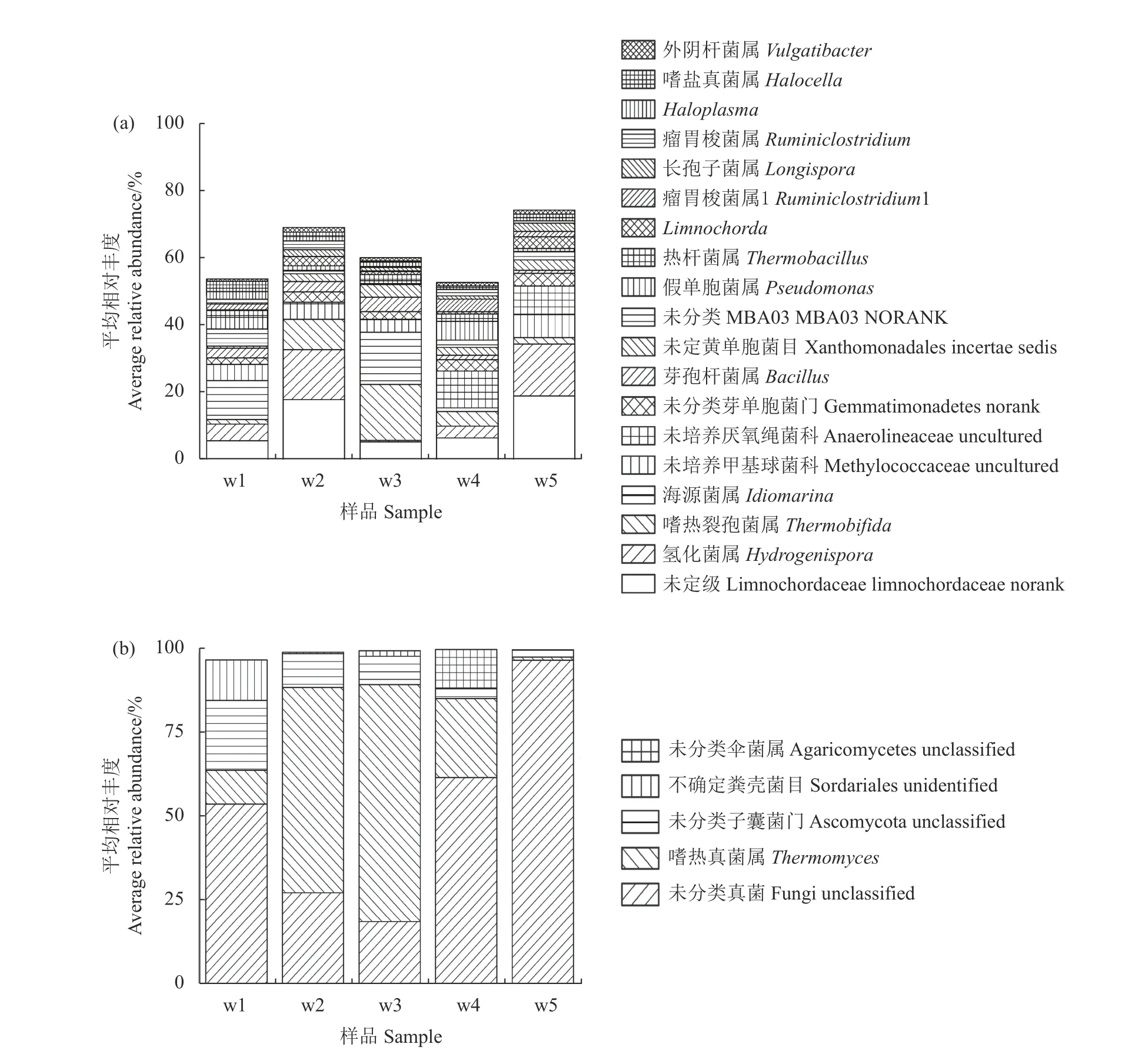
图3 不同时期所取堆肥样品在细菌(a)和真菌(b)属层次上的组成Fig.3 Genera of bacteria (a) and fungi (b) in samples collected at different times
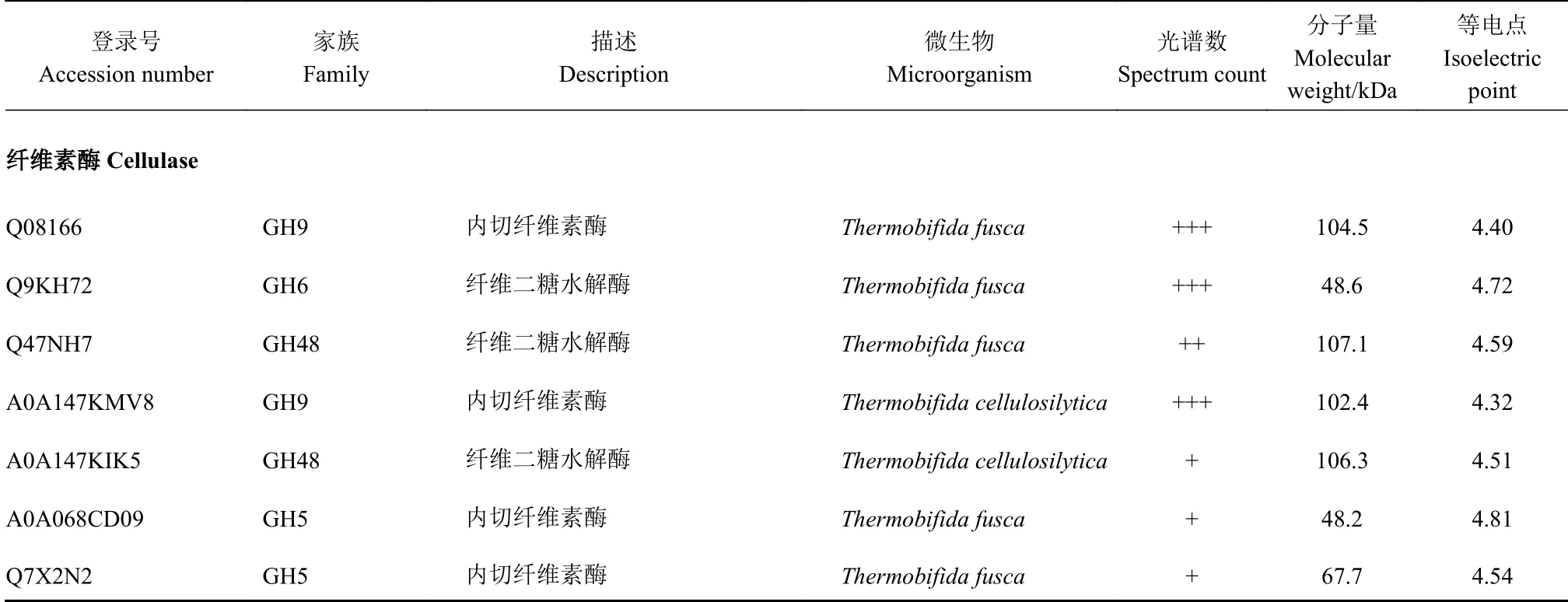
表2 第3 周堆肥样品中主要细菌属和真菌属分泌蛋白的功能鉴定Table 2 Functions of enzymes secreted by dominant bacterial and fungal genera in samples taken in 3rd week

续上表
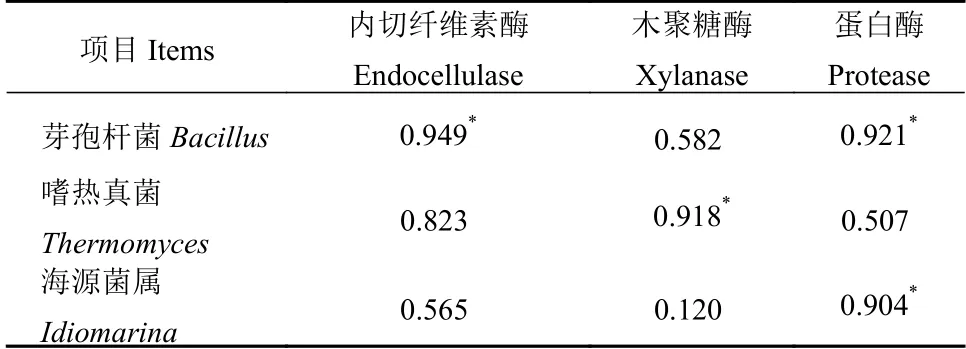
表3 主要微生物属和酶活性的相关性Table 3 Correlations between dominant genera and enzyme activities
3 讨论与结论
本研究利用整合宏组学技术研究了番茄秸秆与玉米秸秆共堆肥生境中的关键微生物及其功能,从基因丰度上看,主要细菌门为放线菌门Actinobacteria(23.1%)、变形菌门Proteobacteria(37.3%)和厚壁菌门Firmicutes(26.5%),主要细菌属为嗜热裂孢菌属Thermobifida(16.5%)、海源菌属Idiomarina(15.6%)、芽孢杆菌Bacillus(4.2%)、黄杆菌属Flavobacterium(2.4%)、热 杆 菌 属Thermobacillus(1.9%)、 糖 单 孢 菌 属Saccharomonospora(1.4%)和清野氏菌Planifilum(1.2%);主要真菌门为子囊菌门Ascomycota(81.3%),代表属为嗜热真菌属Thermomyces(70.5%)。嗜热裂孢菌属Thermobifida 功能多样,既通过产生GH5、GH9 家族的内切纤维素酶,GH6、GH48 家族的纤维二糖水解酶参与纤维素的降解;又通过分泌GH10 家族的β-木聚糖酶参与半纤维素的降解;同时参与果胶裂解。糖单孢菌属Saccharomonospora 主要参与蛋白质的降解,分泌多种丝氨酸蛋白酶和胰酶,同时通过产生GH10 家族的β-木聚糖酶,参与木聚糖的降解。其他功能菌还有,清野氏菌Planifilum 和嗜热真菌属Thermomyces 分别通过产生GH10 和GH11 家族的内切-1,4-β-木聚糖酶参与半纤维素的降解,海源菌属Idiomarina 通过分泌丝氨酸蛋白酶参与蛋白质的降解。
在本研究中,嗜热裂孢菌属Thermobifida 是唯一的纤维素降解者,这与Anbarasan 等[29]的研究结果基本一致,认为嗜热裂孢菌属Thermobifida 因能产生多种纤维素酶和半纤维素酶,是秸秆类堆肥物料的主要降解者。除嗜热裂孢菌属Thermobifida 外,糖单孢菌属Saccharomonospora、清野氏菌Planifilum 和嗜热真菌属Thermomyces 共同参与番茄秸秆半纤维素的降解,而Zhang 等[7]关于玉米秸秆条垛式堆肥的研 究 发 现,Thermomyces lanuginosus、Aspergillus 和Thermopolyspora 是半纤维素的主要降解者。这种功能微生物的差异可能因堆肥物料的组成和性质、环境因子,如温度、湿度、供氧量和C/N 等因素有关[30]。
同时,Liu 等[6]利用DGGE 技术研究了生物炭、泥煤沼和沸石等调理剂对番茄秸秆堆肥过程中细菌群落结构的影响,结果表明黄杆菌属Flavobacterium对应条带较明显,且广泛存在于堆肥发酵各阶段。在本研究结果中,黄杆菌属Flavobacterium 同样具有相对较高的丰度(2.4%),但质谱检测时并未检出源于该菌的蛋白。由于DGGE 指纹图谱技术和高通量焦磷酸测序技术均是基于DNA 的序列差异对微生物的群落结构进行表征。在样品总DNA 的提取过程中,无论是有生命的还是已经死亡的生物体DNA 均会被提取出来。例如,据估算,土壤总DNA 的40%源于已经死亡或不完整的生物体[31]。因此,仅通过分子生物学手段或高通量焦磷酸测序,以基因丰度来表征微生物的重要程度是有挑战性的[32]。
基于蛋白质水平的检测技术(如质谱等)仅对活的细胞及其产生的蛋白质进行分析,其表征的功能微生物群落结构肯定会与基于DNA 水平(如DGGE、高通量测序等)的微生物群落结构有所差异。如在本研究中,Flavobacterium 的基因丰度虽占细菌全部序列的2.4%,但却没有检测到对应的胞外蛋白。因此,只有将高通量焦磷酸测序和宏蛋白质组学方法联合起来,形成“整合宏组学方法”,才能更好地将微生物的群落结构及其参与的生态过程联系起来。
此外,本文仅检测了植物生物质和蛋白质降解最旺盛时期(第3 周)的微生物群落组成及关键微生物的功能。整个发酵周期内主要微生物的群落演替及其功能变化需要进一步深入研究[33]。
