介尺度视角下的电催化剂调控策略
2020-10-27郑星群李莉魏子栋
郑星群,李莉,魏子栋
(重庆大学化学化工学院,重庆401331)
引 言
电化学催化是化学能转化过程中的核心,是催化化学与电化学密切结合的产物。电化学催化除了具有异相催化的全部特征之外,还可以改变电极电势实现反应方向和反应速度的调控与反转。例如,电极电势通过调变电极上电子的能量状态,不仅可以改变电极反应的活化能,还影响着物种在电极表面的吸附状态和吸附强度,因而影响着电极反应的动力学。虽然外加电极电势可实现许多困难的催化反应,但如何最大限度地降低过电位、节能降耗、提高催化效率,仍是电化学催化面临的主要挑战。电催化剂作为电化学催化反应中的核心,其活性位结构、密度、本征活性、稳定性以及催化剂的导电性决定着电催化性能的优劣。据此,如何有效地构筑和调控催化剂表界面的活性位,同步提高导电性、活性与稳定性,成为电催化剂设计和优化的关键和挑战。
催化剂掺杂、引入空位、多组分的复合、调变载体等是提升催化性能较常用的策略[1−5]。这些方法看似各不相同,但从介尺度的视角可以发现它们都具有相同的调控机制和思路。当某催化剂引入掺杂原子、空位以及第二相材料时,其结构、组成与性质等从A 到B 的变化过程中,介尺度区域内往往会出现区别于两者极端情况条件下的第三种主导性质——“介尺度性质”。如图1 所示,该性质是由于催化剂表界面从组分Ⅰ到组分Ⅱ变化时,组分间因几何、电子结构的差异引起电荷转移、缺陷、晶格位错、应力变化甚至晶体结构突变等因素,导致介尺度区域具有与两个极端情况截然不同的介尺度性质,如电子结构,从而呈现不同的催化性能。这种某一因素与其调变性质间的非线性关系称为“介尺度现象”。而该现象存在于各类金属、金属化合物以及多相复合催化剂的构筑与制备中。据此,本文秉持“介尺度”学术思想,对近年不同催化剂表界面活性位构筑与调控策略中存在的介尺度现象及其对应的介尺度性质进行了总结,主要包括:晶体结构、化学组分、相界面以及应变效应对催化剂结构和性能的调变。
1 催化剂晶体结构的调变
无机催化材料因多变的晶体结构而具有不同的原子排布和电子结构,从而表现出不同的物化性质和催化性能。通过物理、化学手段对催化剂晶体结构进行调变是催化剂设计及调控的常用策略之一。
对金属基催化剂,特别是一些过渡金属基电催化剂,因其原子堆积方式不同往往表现出不同的催化活性。Kusada 等[6]发现,与hcp−Ru 纳米粒子相比,密堆积的fcc−Ru 纳米粒子表面暴露了更多的活性位点,从而对CO 氧化具有更高的催化活性[图2(a)]。Wang 等[7]以Ni 为例探究了不同晶体结构的纳米颗粒对氧析出反应(OER)的影响,发现具有相似尺寸和氮掺杂碳外壳(NC)的hcp−Ni较fcc−Ni具有更高的OER 活性。这是由于内部hcp−Ni 金属的电子较fcc−Ni 更易隧穿至外层NC,增加Fermi 能级处的态密度从而调节NC 表面的电子态,促进NC 表面的OER 反应。同理,他们发现hcp−NiFe 合金纳米颗粒也比fcc−NiFe 合金纳米颗粒更有利于提升OER 催化活性[8]。此外,Tong 等[9]成功制备了有序的bcc−PdCu合金和fcc−PdCu 合金。他们发现晶相从fcc到bcc 的转变使得PdCu 对N2还原中间物种的吸附增强而对H 的吸附减弱,从而提升N2还原反应(NRR)的催化活性和选择性。

图1 电催化剂表界面性质调控的介尺度现象Fig.1 Mesoscale characteristics encountered when tuning surface/interface properties of the electrocatalysts
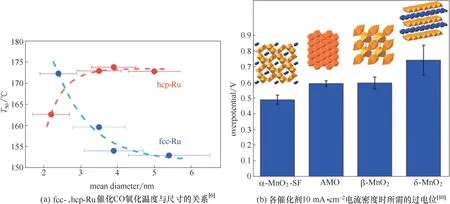
图2 不同晶体结构催化剂的活性比较Fig.2 Comparison of the activities of catalysts with different crystal structures
过渡金属化合物,特别是氧化物和硫属化合物,因具有复杂多变的晶体结构而在电催化领域得到广泛的应用。如Meng 等[10]合成了α−,β−,δ−和非晶态MnO2(AMO)并发现其氧还原OER 活性遵循α−MnO2>AMO>β−MnO2>δ−MnO2的顺序[图2(b)]。α−MnO2由于其独特的(2×2 通道)结构,使得其相比于β−MnO2(1×1 通道)和δ−MnO2(层状结构)具有更大的孔隙结构,不仅可以暴露出更多的活性位点还可以使反应物更好地传输到催化活性位点之上进行反应。此外,α−MnO2中含有阳离子(K+)和水还可使其晶胞更高概率地暴露在固液界面上,从而使晶体结构内部的金属活性位点也得以被利用。Yang 等[11]发现MnO2催化甲苯氧化活性也与晶体结构密切相关,其活性顺序为δ−MnO2>α−MnO2>γ−MnO2>β−MnO2。δ−MnO2具有更高活性的原因则是由于通道中K+使晶体结构发生扭曲更易产生氧空位,从而利于甲基的快速脱氢。Xie 等[12]通过相转变策略实现了斜方相CoSe2(o−CoSe2)和立方相CoSe2(c−CoSe2)的可控合成,实验及DFT 计算表明c−CoSe2较o−CoSe2具有更高的导电性,更适中的H 吸附自由能,以及优异的析氢(HER)活性。
基于晶体结构对电催化性能的影响现象,可利用“介尺度”的思想,调变晶体结构从一个极端到另一个极端,探究晶体结构与催化剂性能间构效关系的同时实现催化剂性能的最大化。Jaramillo 研究团队[13]研究了IrOx及其混合氧化物晶相变化与其OER活性间的关系。他们通过调变Ir 基氧化物中Sr 的含量,制备出三种纯相氧化物(Sr4IrO6, Sr2IrO4以及SrIrO3)。各催化剂的OER 本征活性顺序为IrOx<SrIrO3<Sr4IrO6<Sr2IrO4,表明Ir∶Sr 比例适中时的晶相Sr2IrO4具有最优的催化活性。过渡金属尖晶石结构氧化物的组成非常复杂,其可以用(A1λBλ)四面体位点[AλB2λ]八面体位点X4公式精准描述[14]。随着尖晶石结构中的λ 的变化,其结构也会从正式结构(λ=0)转变为复杂结构(0<λ<1),最后转变为反式结构(λ=1)。对比于正式结构,反式结构的八面体位点中包含两种元素,其不同的组成可能对其氧还原(ORR)催化性能有影响。Wei 等[15−17]通过调节尖晶石中八面体体心位Fe 元素的浓度(从无到有),尖晶石结构经历了“正尖晶石−反尖晶石−正尖晶石”的结构循环。他们发现具有反尖晶石结构的CoⅡ(FeCo)ⅢO4具有优于Pt/C 的ORR 催化活性(图3)。这源于随Fe 含量的增加, 尖晶石从半导体向半金属转变, 即处于介尺度区域的反尖晶石型{Co}[FeCo]O4呈现半金属性,具有更好的导电性。此外,反尖晶石型{Co}[FeCo]O4因结构反转促使居于八面体体心的Fe、Co间出现明显的电荷转移,使Fe 富电子而Co 缺电子,Fe−Co 元素间的电荷差异称为电荷“异化效应”。该效应不仅促进了O—O键的活化还利于OH的脱附(速控步)从而提高了ORR 活性。Cheng 等[18]也发现Co−Mn−O 尖晶石ORR 催化性能与其晶相结构有关。在ORR 测试中,具有立方晶系结构的Co−Mn−O 尖晶石(CoMn−P)的ORR 催化活性明显高于具有正方晶系结构的Co−Mn−O 尖晶石(CoMn−B)。通过XPS、TPD以及DFT 计算分析发现,由于两者不同的晶体结构,使得氧气在其表面上的吸附能力不同,即在CoMn−P 催化剂上,氧气的吸附能力明显强于CoMn−B从而有利于O2的进一步还原。

图3 晶体结构反转诱导的电荷异化效应及各尖晶石催化剂的ORR催化活性对比[15−17]Fig.3 Dissimilarity effect resulted from reversing the crystalline structure and comparison of the ORR performances of these catalysts [15−17]
2 催化剂表界面化学组分的调变
催化剂表界面化学组分的调变也是有效调节催化剂性能的常用策略。化学组分的改变常源于元素种类和比例的改变,如通过引入空位、掺杂原子等方法调变催化剂组分中阴/阳离子的种类、含量、比例等。而这些变化对催化剂活性及稳定性的调变往往也表现出介尺度现象,即当元素种类、比例等处于介尺度区域时往往较未调变前的极端情况具有更高导电性、稳定性和催化活性。
金属氧化物中氧空位的变化对催化剂导电性和活性具有显著的影响。Cheng 等[19]报道发现MnO2可以通过简单的热处理即可引入氧空位(OVs)缺陷,提升其作为ORR 催化剂的活性。进一步地,他们将MnO2在氢气气氛下煅烧一段时间发现,其电导率从原来的7.81×10−2S·m−1增加到0.26 S·m−1,即与没有经过氢气还原热处理的MnO2相比,热处理后得到的MnO2具有更好的导电性[20]。Wei 等[21−22]早前研究表明直接热解硝酸锰制备MnO2时,适当提高热解温度或者在合成过程中额外加入Mn3O4,均会提高其催化ORR 活性,且该活性与晶面距d 为2.72 Å(1Å=0.1 nm)的晶面密切相关。后期研究发现该晶面的形成正好与晶体中的氧空位有关。β−MnO2中氧空位的数目增加,d=2.72Å 晶面的衍射峰强度也随之增加,证实该特定晶面由氧空位诱发晶格收缩所致[23]。进一步,DFT 计算发现MnO2的ORR 活性与氧空位浓度呈现出非线性关系,证实氧空位浓度调变其催化性能的确存在介尺度现象,即适中氧空位浓度可获得更优的导电性和催化活性。能带结构变化表明,随着氧空位浓度的增加,β−MnO2带隙先变窄再变宽。当氧空位数量增加到12 时,β−MnO2−12OVs 显示出最窄的带隙宽度,即拥有最好的导电性。与此同时,如图4所示,催化剂的Fermi能级、活化氧气的O—O 键长,也表现出了类似于导电性的变化规律,证实适当浓度的氧空位可有效提高MnO2催化剂的导电性和Fermi 能级,从而利于氧气与催化剂之间轨道的重叠和电子转移;氧空位还可促使MnO2晶体发生Jahn−Teller 效应,诱发MnO2表面Mn−Mn 键收缩或拉伸,使活性中心与O—O 键长呈最优匹配,利于O2的活化离解。该研究结果也得到其他独立研究团队的证实[24]。同样地,其他类型氧化物,如CaMnO3[25]、NdNiO3[26]、MnCo3O4[27]等的导电性和电催化活性也在适当的氧空位浓度时具有最优值,证实了适当氧空位浓度下的催化活性具有介尺度性质。

图4 氧空位浓度对MnO2活性与导电性调节的介尺度现象[23]Fig.4 Mesoscale relationships between concentration of oxygen vacancies and MnO2 activities and conductivities[23]
金属元素或阳离子种类和比例的变化,可以有效调节其配位环境和化学价态从而进一步优化其催化性能。如钙钛矿金属氧化物中同时存在A 和B位,调节A、B 位上的元素组成以及比例可使得其中B 位点上的过渡金属元素的eg轨道的电子数接近1,进而具有最佳的氧还原催化活性。Luo 等[28]通过引入A 位缺陷并同时对B 位点进行Ir 掺杂,改变了(La0.8Sr0.2)1−xMn1−xIrxO3的结构,有效提高了该钙钛矿氧化物的氧还原催化活性。Cheng等[29]发现,立方晶系结构的Co−Mn−O尖晶石,不同的Co∶Mn元素比也会影响氧气分子的活化程度以及活性位数目,优化后的CoMn−P催化剂具有与Pt/C催化剂相当的ORR活性。掺杂阳离子同样会诱发催化剂化学价态与性质的变化。如Xu等[30]通过溶胶−凝胶法制备了一系列Li+掺杂的Ni1−xLixO,并研究了不同Li+掺杂量对NiO 结构及催化CO 氧化活性的影响。他们发现Li+掺杂量较低时(x<0.16),Li+取代晶格中的Ni2+并形成固溶体结构,随着掺杂量增加,固溶体中Ni3+含量增加从而诱导大量流动氧的形成使其催化CO 氧化的活性逐渐提高,且当x=0.10 时,催化剂具有最高的CO 氧化活性;但随着Li+掺杂进一步增加(x>0.16),NiO 表面逐渐形成Li2CO3,阻碍Ni3+的形成并覆盖活性位点,使其催化活性降低。
与金属元素/阳离子的组成变化相比,非金属元素,如阴离子配体的改变可在更大范围内调变金属化合物催化剂的活性与稳定性。不同阴离子形成的化合物往往具有截然不同的晶体结构、物化性质与催化性能。通过改变配体阴离子的组成以及比例,极有可能在两极端单一配位之间的介尺度区域内获得意想不到的催化性能。例如,Co 的硫化物(Co3S4)[31]具有较高的OER 活性,但因S容易流失而稳定性较差;Co 的氢氧化物(Co(OH)x)[32]虽具有较高的稳定性,但其OER 活性差。当Co 化合物兼具OH 与S2−配体时,其是否同时具有较高的活性与稳定性,基于此思想,Wei 课题组[33]利用水热法和二次水热硫化实现了前体NiCo2(OH)2中的OH 配体和S2−的交换反应,并得到双配体催化剂NiCo2(SOH)x。他们发现具有介尺度配体结构的NiCo2(SOH)x兼具了两极端组分NiCo2(OH)x的稳定性与NiCo2S4的活性,实现了催化剂活性与稳定性的同步提升。如图5 所示,这主要源于双配体化合物表面的OH 和S 协同调控了催化剂的电子结构和化学环境,使介尺度区域的NiCo2(SOH)x消除了自身磁性,从而减弱了含氧物种的吸附;M−S 反键轨道的部分电子转移到M−O 反键轨道,在减弱M−O 键的同时,大大地加强了M−S键,从而抑制了催化过程中S 原子的流失,提高了稳定性。研究发现双配体化合物(NiCo2(SOH)x)较单阴离子配体的氢氧化物、硫化物,以及商业RuO2催化剂具有更高的OER 活性和稳定性。Zhang 等[34]结合化学气相生长和Li−插层法制备了一系列1T’相ReS2xSe2(1x)(x=0~1)纳米点(NDs)。电化学测试表明,S∶Se处于中间比例时,即ReSSe,催化剂具有最高的HER 电催化活性,且其活性远高于两极端ReS2和ReSe2催化剂。他们发现ReSSe NDs中Re−S和Re−Se键具有不饱和配位,配位数分别为~1.4 (Re−S)和~1.9(Re−Se),其在边缘位置和S 空位处暴露更多的Se 原子。进一步DFT 计算表明这些不饱和配位的Re原子以及边缘处的Se原子正是HER活性位点。

图5 双阴离子配体协同调控的介尺度现象[33]Fig.5 Mesoscale characteristics tuned by the dual−ligand sytnergistic modulation[33]
除金属化合物催化剂外,碳基类催化剂表界面掺杂原子对其性能调变也具有介尺度性质。Wei团队[35]采用聚二胺吡啶(PDAP)为氮碳前体,再通过氢键在PDAP 表面原位自组装的植酸超分子集合体(PPSA)为磷源,直接热解合成PNC 催化剂。PPSA 的引入实现了氮碳材料表面原位掺P和表面碳缺陷结构构筑,诱导并促进N 杂原子掺入碳环结构,形成N/P 比为1∶1 的P−N 共掺杂结构。掺杂过程表现出介尺度现象,如图6 所示,仅适当的P 含量可形成结构稳定的P−N 共掺杂结构,过量的P 含量则使N 由六元环N 向五元环转换,而由于P 原子更倾向结合O,从而破坏P−N 共掺杂结构。N 和P共同掺杂结构会使得邻近的碳原子的电子密度重新分配,强化氧气在催化剂表面的选择性吸附,显著降低NOx和SOx在催化剂表面的富集程度。实验证实PNC 催化剂在含有SO、N和HPO的模拟毒化环境下,PNC催化剂ORR催化活性几乎未有衰减,说明PNC催化剂对SOx、NOx和POx有良好的抗中毒性能。而相同环境下,Pt/C、NC 和PC 催化剂出现不同程度的中毒特征,ORR催化活性呈现不可逆的衰减。
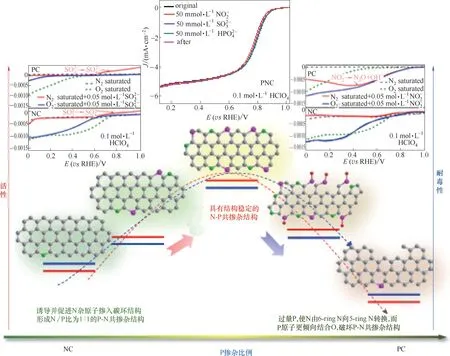
图6 N,P共掺杂碳基催化剂活性与抗毒性调节的介尺度现象[35]Fig.6 Mesoscale properties of activity and anti−toxicity regulation of N,P co−doped carbon−based catalysts[35]
3 催化剂相界面诱导的介尺度性质
大量研究[36−38]证实,采用化学方法制备的复合催化剂往往较简单物理混合具有更高的催化性能,其根本原因是各组分之间形成了存在一定化学键合的相界面。各组分相界面间化学键合方式、组分间的强相互作用、电荷转移以及晶格不匹配产生的应变作用等均会诱导相应的复合催化剂展现出截然不同的催化性能,这也势必导致介尺度区域内的异相界面区域呈现出介尺度性质。
金属/金属化合物复合催化剂是目前研究较多的一类复合电催化剂[39]。金属−化合物间的化学键合产生的强相互作用可导致各组分具有与纯组分截然不同的几何电子结构,从而使其呈现出较纯组分更优的催化活性。Yu 等[40]报道了多孔碳载Ni/Mo2C (Ni/Mo2C−PC) 复合催化剂相较于单一的Ni−PC、Mo2C−PC 催化剂以及物理混合的Mo2C−PC 和Ni−PC 催化剂在碱性介质中具有更高的HER 和OER 电催化活性。Ni/Mo2C−PC 的高活性源于Ni 转移部分电子给Mo2C,使Ni 的价态更高而Mo 的价态更低,从而提供了更多的有效活性位点。Villullas等[41]发现相同尺寸的Pt颗粒与不同过渡金属氧化物载体间的电荷转移直接影响了Pt 5d 空穴,并且Pt 5d 空穴数与ORR 电流密度之间存在线性关系。此外, 通过化学方法调节载体的表面状态,如表面组成、空位等,也可调节金属/氧化物间电子相互作用。如Gong 等[42]发现TiO2载体的表面状态能明显影响Pt 的催化性能。他们在沉积Pt 催化剂之前,通过在不同的气氛中对载体进行预处理,得到了氧化型、羟基化型、还原型的TiO2载体(Pt/o−TiO2, Pt/h−TiO2和Pt/r−TiO2)。这些不同表面状态的TiO2不仅使Pt分散性出现差异,还使Pt−TiO2之间的电子转移显著不同。相较于Pt/o−TiO2和Pt/h−TiO2,Pt/r−TiO2上的Pt 颗粒分散度最小,界面处Pt 具有最小的正电荷,且催化CO 氧化反应的能垒最低,因此具有最高的CO氧化反应活性。
进一步研究发现,复合催化剂各组分间的强相互作用和电荷转移主要对各组分相界面间区域的几何、电子结构产生显著影响,致使相界面成为决定催化剂活性的关键。Cheng 等[43]通过高压磷化法制备了含Ru−Ru2P 界面的Ru/Ru2P 碱性HER 催化剂。他们认为Ru 与Ru2P 复合所形成的界面不仅提供了H2O 解离的位点还通过调节Ru2P 的电子结构使其具有更佳的H 吸附自由能。Wang 等[44]制备出了含有Ni/NiO 界面的NiOx@BCNTs 催化剂。实验与DFT 计算证实本体金属Ni0与Ni/NiO 界面金属Ni0具有较高的HER 本征活性,其中界面Ni0较本体Ni0具有更高的催化活性,揭示了界面在催化中的重要作用。Wei课题组[45]进一步证实金属氧化物/金属相界面间的金属具有与本体金属和氧化物截然不同性质,如Ni/NiO 界面的形成导致界面Ni 较纯Ni 出现了晶格膨胀,同时具有不同的化学价态。DFT 与实验证实金属氧化物/金属相界面间的电荷转移,导致界面金属与本体金属间的电子结构出现异化特征,即沿着“氧化物/金属”界面形成了对OH 和H2O具有排斥作用,而对H 具有最优选择性吸附的界面通道,该界面通道随即成为专属的形成H 原子复合并释放H2的通道,如同在“氧化物/金属”界面周围形成H2快速抽提的“烟囱”,称之为金属/金属氧化物界面对HER 的“烟囱效应”[图7(a)]。显然,金属氧化物与金属之间的界面含量越大,沿界面形成的烟囱越多,催化剂的活性越高[图7(b)]。Hu课题组[46]利用Ni纳米晶自然氧化的尺寸相关性来精确调控纳米晶表面Ni/NiO 比例的方法,成功制备得到一系列Ni/NiO 纳米异质界面可调的高度分散的Ni/NiO 纳米晶模型催化剂,进一步证实了碱性介质中HER 活性与Ni/NiO 异相界面组成的相关性。他们发现平均尺寸为3.8 nm 的Ni/NiO 纳米晶具有最佳的Ni/NiO 比值(~20%),从而具有最优的HER 活性和稳定性。同样地,复合催化剂相界面对催化氢氧化(HOR)也存在“烟囱效应”[图7(a)]。Wei 课题组[47]发现将Ru 簇金属负载于TiO2纳米片时,可通过Ru−TiO2界面形成的Ru−O 键调节界面Ru 簇的氧化程度[图7(c)]。界面Ru 簇的氧化程度与其表面OH 的吸附强度呈反比。当界面氧化的Ru 的4d 轨道达到半充满状态的同时又保持了表面金属特征,使其在催化HOR 过程中免于吸附H2O 和OH 而被氧化,仅选择性利于H2的离解与催化氧化。Sun 等[48]也证明Ni/NiO 在界面处拥有更加适中的H 吸附能从而协同促进HOR反应。
金属/氧化物界面间的结合方式会进一步调变界面区域的晶格和电子结构。如图8 所示,Wei 课题组[49]报道了一种金属团簇嵌入氧化物的“晶格限域型”Ru@TiO2催化剂。还原气氛下,在无定形TiO2的晶化过程中,Ru纳米粒子在缺陷位处沿TiO2晶格中Ti 原子生长成簇,形成以Ru−Ti 金属键连接的Ru@TiO2晶格结构。这种金属键合的晶格限域结构,导致界面的Ru,一方面具有类似与TiO2氧化物的原子排列结构(Ru 原子按TiO2(101)排列),同时保持了其固有的金属性;另一方面,又呈现出既异于Ru 块体金属又区别于Ru 纳米团簇的介尺度性质,即同时具有块体的金属性和团簇的低配位性(CN=7~8);具有如团簇金属的反应活性和块体金属的高稳定性;具有大于金属氧键(Ti−O,0.193 nm)却低于金 属 键 的(Ru−Ru,0.267 nm)的Ru−Ti 键 长(0.258 nm)。这种具有介尺度性质的Ru@TiO2催化剂既不同于传统“负载型”催化剂,又不同于完全包覆的“核⊙壳型”催化剂。Ru−Ti 界面金属键促进电子从富电子的n 型半导体TiO2转移到Ru 簇中的d 带上,使Ru 簇的d 带接近完全充满的状态。这种电子几乎全满的d 带,有利于维持Ru 的金属性,提高其催化氢氧化反应活性同时,大幅减少了CO的σ电子向Ru 金属d 空轨道转移的概率,削弱了CO 吸附强度,弱化了CO 中毒,从而赋予该催化剂超优异的抗CO中毒性能。在含量0.1%的CO 存在的条件下,氢氧化催化活性几乎不受干扰;甚至CO 含量提高到约10%(体积分数),该催化剂仍选择性催化氢氧化。此外,在d 带电子几乎全满的Ru 簇表面,Ru 更趋稳定,不易氧化,使该催化剂在较宽的电位范围仍保持反应活性。在酸性和碱性介质中,Ru@TiO2催化剂HOR 质量活性(@0.02 V)较商业化PtRu/C 催化剂高出15%~30%;其超强的抗氧化性,在电位高达0.9 V 时(一般金属都会以表面生成氧化物种为主)Ru@TiO2催化剂中的Ru 簇仍保持其表面的金属性和催化氢氧化性能。
上述金属/金属氧化物界面的介尺度性质也存在于化合物/化合物[50],晶相/无定形相等各类含有相界面的复合催化剂[51]中。如Wei 课题组[50]构筑了含NiO−Ni3S2异质结界面的高效双功能HER/OER 催化电极(图9)。他们发现Ni3S2和NiO 之间的相互作用和界面电荷转移,导致界面Ni电子组态的变化,Ni−S 键结合能增强而Ni−O 键结合能降低。同时,NiO−Ni3S2/NF 的HER 和OER 催化活性均大于单组分NiO/NF和Ni3S2/NF催化剂活性之和,说明NiO−Ni3S2/NF 的高活性不仅是单组分NiO/NF 和Ni3S2/NF 催化剂作用的叠加,更是因为Ni3S2−NiO之间相互作用产生了高活性界面区。此外,调控Ni3S2和NiO 的比例发现活性界面区含量受组分的相对含量影响,活性界面区域越大,NiO−Ni3S2/NF 的催化性能就越高。Song 等[52]通过表面F 化处理得到了含大量晶相−无定形相界面(c−a)的CoB2催化剂。他们发现该催化剂的OER 性能较纯相CoB2有显著提升,当过电位为320 mV 时TOF 增加了40 倍,法拉第效率为99.9%。理论和DFT 计算证明在c−a 界面上,无定形相为氢氧化物或水分子提供大量的吸附位点,且其部分氧化降低了OER 基元反应能垒,同时增加了载流子浓度。与此同时,晶相CoB2可以促进吸附物种和催化剂之间的电荷转移,从而晶体和非晶相c−a 界面密度的增加可以显著增强OER活性。
按照上述思想,复合多组分构筑具有更多相界面的复合催化剂可使催化剂具有更优的催化活性。Zhang 等[53]报道构筑含有NiO、Ru 以及多孔Ni 的三相界面的复合催化剂具有较双组分催化剂具有更强的协同增强效应,可同步提升HER中的Volmer和Heyrosky基元步骤,其HER的过电位仅39 mV@10 MA·cm−2,优于非Pt 催化剂。Wang 等[54]报道了一种镶嵌于孔状碳阵列的Co/CoO/Co3O4异质结催化剂。该催化剂中Co的存在提高了导电性,具有结构缺陷和氧空位的纳米级CoO/Co3O4异质结增强了界面的电荷转移并促进了O2的吸附,从而加速了ORR动力学,降低了反应能垒。
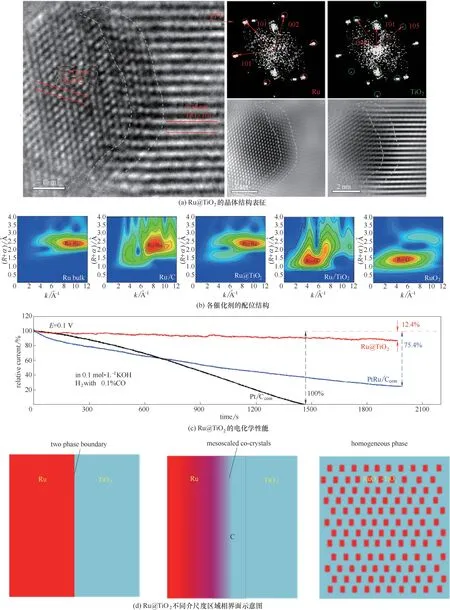
图8 晶格限域Ru@TiO2催化剂的结构及电化学性能[49]Fig.8 Structure and electrochemical performance of lattice−confined Ru@TiO2[49]
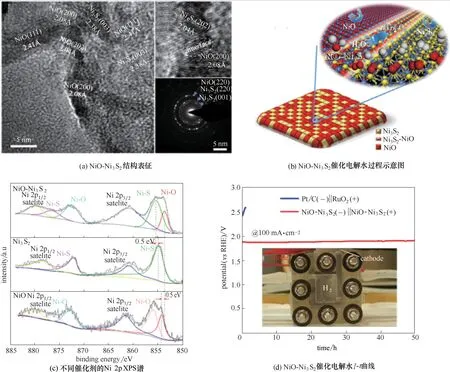
图9 NiO−Ni3S2/NF催化电极结构及OER活性[50]Fig.9 Structure of NiO−Ni3S2/NF electrodes and their activities toward OER [50]
4 应变效应对催化性能的诱变
在构筑多相催化剂时不仅会引入因不同组分间电子相互作用产生的配位效应,还会引入因晶格失配或键长变化而产生的应变效应[55−56]。此外,对于单相催化剂,各类缺陷(掺杂、空位、位错)的形成及形貌结构的变化也会引入不同程度的应变效应和配位效应。配位效应的作用往往存在于1~3 个原子层间,是一种短程效应。而应变效应是一种长程效应,其影响可存在于1~6 个原子层间,从而对特定结构的催化剂,如核壳结构金属催化剂、金属覆盖层型复合催化剂等的几何电子结构及催化性能具有显著的影响[57−59]。因此引入或调节应变效应作为一种有效的调控策略而被广泛应用于催化剂,特别是金属基催化剂的设计及表界面活性的优化。

图10 不同应变对催化活性的影响[67,69]Fig.10 Effects of different strains on catalytic activity[67,69]
基于d 带中心理论的应变效应[57−58,60−62]表明:拉伸应变可以使后过渡金属(d轨道半充满及以上的金属)的d 带中心上移(靠近Fermi 能级),增强金属−吸附物之间的(M−ads)相互作用;压缩应变使d 带中心下移(远离Fermi 能级)削弱M−ads 相互作用,即应变效应与d带中心呈线性关系。对于需要减弱中间物种吸附的催化反应,引入压缩应变可弱化物种在金属基催化剂表面的吸附,提高催化活性。以ORR 为例,诸多研究[63−66]也证实,压缩应变有助于提升Pt 等贵金属基催化剂表面的ORR 活性。如Wang 等[64]通过DFT 计算证明由于压缩应变引起的d 带变宽使d带中心负移,ORR 物种在压缩应变的Pt(111)表面上的结合能比在非应变表面上的结合能更弱,且压缩应变下Pt(111)面的ORR 决速步活化能更低,即压缩应变有助于Pt(111)面ORR 活性的提升。Bard 等[67]通过实验考察了应变效应对Pt ORR 活性的影响。他们利用双向形状记忆效应,以NiTi 形状记忆合金为基底,通过不同的热循环处理,使沉积在表面的Pt 纳米薄膜得到了三种不同的应变状态:无应变、压缩应变和拉伸应变状态。研究发现,与无应变Pt膜相比,具有压缩应变的Pt膜表现出更高的ORR活性,拉伸应变Pt膜的活性最差[图10(a)、(b)]。具体表现为,压缩应变使Pt 膜上Hupd和OHad的覆盖度显著降低,其ORR 动力学速率常数增加了52%,半波电位正移了27 mV;拉伸应变Pt 膜催化ORR 动力学速率常数降低了35%,半波电位出现了26 mV的负移。证实了压缩应变和拉伸应变对ORR活性的影响与d带中心理论的预测一致。Wang 等[68]也发现Pt 表面分别被压缩和拉伸5%时,其ORR 活性分别增加90%和降低40%。
尽管许多研究证实了应变效应对ORR 活性的影响与d 带中心理论的预测一致,即拉伸应变会增强物种吸附,但近年来的一些研究[69−71]发现拉伸应变也存在弱化中间物种吸附强度的现象。Yang等[71]通过DFT计算发现在Pt(100)面拉伸应变可以减弱CO 和O 的吸附并降低CO 氧化的能垒从而促进CO 氧化反应。Adzic 等[72]将单层Pt 沉积到PtPb、PdPb 和PdFe 金属间化合物纳米颗粒上,其中Pt/PdFe上Pt被过度压缩,Pt/PdPb上Pt被过度拉伸,Pt/PtPd 上Pt 则适当被拉伸。这些催化剂的ORR 活性顺序为Pt/PtPd>Pt/PdFe>Pt/PdPb,说明具有一定拉伸应变的Pt 原子层也可以提高ORR 活性。但值得注意的是,因为Pt 原子层厚度小于0.6nm(约3 个原子层)时,来自基底的配位效应无法忽略[73−74],此时应变效应和配位效应共同决定着表层Pt 的催化活性。为此,Guo等[69]制备了Pt壳约1 nm厚的PtPb/Pt核/壳纳米板催化剂,其Pt 壳约4 个原子层厚度,可忽略PtPb 核对表层Pt 的配位效应,从而更好地评估应变效应对催化活性的影响。他们发现因PtPb 核与Pt壳的晶格失配使该催化剂暴露最多的Pt(110)面具有较大双轴拉伸应变(>7.5%),且这种由PtPb核对Pt壳造成的拉伸应变使得Pt 的ORR 比活性远高于商业Pt/C(约33.9 倍)[图10(c)、(d)]。进一步的DFT 计算也证实在Pt(110)面上适当的拉伸应变有助于将Pt−O结合强度降低到最佳值,从而提高其ORR 活性。Berlinguette 等[75]通过机械给Pd 电极施加拉伸应力也发现Pd 晶格中对H 的吸收量可减少1.1%±0.4%(2%的应变),其电催化HER 活性可增加5.7% ±1.7%(4.5%的应变),从而证明了拉伸应变同样可以弱化物种吸附强度。
显然,金属基催化剂中应变效应对其电子结构的调变不是线性关系,即压缩应变和拉伸应变都可能弱化物种吸附,提升活性,呈现非线性关系(图11)。基于拉伸应变效应有悖于d 带中心理论的预测,且存在异常吸附行为的现象,理清应变效应对电子结构的影响关系,可更好地认识应变效应调变催化活性的介尺度性质。Wei 课题组[76]以Pt 低指数晶面[即Pt(111),Pt(100)和Pt(110)]为例,通过DFT 计算发现,在Pt(111)和Pt(100)应变对表面性质及催化活性的影响与d 带中心理论的预测一致;而对于Pt(110),其εd—5d 轨道中心以及物种的吸附能随表面应变出现非线性变化。如图12 所示,Pt(110)面上拉伸应力一方面会明显增大层内原子距离从而减小原子间轴向dx2−y2轨道的重叠使dx2−y2轨道中心上移,另一方面却缩短层间原子距离从而增加层间dz2轨道的重叠使dz2轨道下移。在压缩应力范围内,dyz和dxz主导了铂−吸附原子间的相互作用,且变化微弱;在拉伸应力范围内,dz2在减弱原子吸附中的作用随应力逐渐增强。同理,其他5d 轨道中心(dxy,dxz,dyz)也表现出类似的趋势。对开放的低原子密度Pt(110)表面随应变表现出异常吸附行为,主要源于表面原子与层间原子键长变化不一致所导致的5个d轨道中心的不一致变化。

图11 金属催化剂应变效应与d带中心、物种吸附的非线性关系Fig.11 Schematic diagram of the non−liner relationship among strain,d−band center and species adsorption energy of metal catalyst

图12 应变诱导的Pt(110)面上物种吸附能、d带中心及各5d轨道中心的变化[76]Fig.12 Changes in adsorption energy,d−band center and the five 5d−orbital centers with surface strain for Pt(110) [76]
除金属基催化剂外,应变效应还广泛应用于一些金属化合物和非金属催化剂的活性调控中。如Rappe 等[77]结合DFT 计算和机器学习发现对于不同浓度及种类的非金属掺杂的Ni3P2,其Ni3−hollow 位的Ni−Ni 键长可以作为HER 活性描述符,这表明非金属掺杂剂对Ni3−hollow 位点产生了一种化学应变效应,可通过压缩和拉伸改变其反应活性。Xue等[78]发现拉伸应变有助于Ni(OH)2纳米带暴露4配位的Ni 活性位点从而促进了OER 活性。Liu 等[79]发现生长于单晶SrTiO3基底上的La0.7Sr0.3CoO3−δ薄膜,因存在压缩应变,可有效降低O 空位浓度从而提高OER活性。Terakura 等[80]证明拉伸应变可以打破ORR 中间物种在N掺杂石墨烯上的吸附能比例关系。
以上结果均证明应变是一种有效调控催化剂活性的策略,应变与活性间的调变关系具有介尺度现象。在催化剂设计中,可结合催化材料的本征性质(如轨道重叠特征、晶面特征、对中间物种的吸附性质等)通过化学方法[57−58,60,81−82](如调节催化剂的组分、复合形式、表/界面结构或形貌等)或物理方法[60](如施加或改变外力、电、磁、温度等)引入或调控应变效应从而对催化剂表界面活性进行调控。
5 结 论
综上所述,介尺度现象和介尺度性质存在于电化学催化剂的设计、构筑与制备等各个方面。催化剂表界面晶体结构、晶面、组分种类和比例、相界面之间的相互作用;各组分、相界面之间的复合成键方式、电荷转移以及应变效应等均可促使催化剂表界面几何电子结构呈现非均一性的特点,从而使其催化性能呈现出非线性变化的介尺度现象。正是这种介尺度现象,促使处于中间状态的介尺度区域往往呈现出与极端情况截然不同的介尺度性质。换句话说,可利用介尺度区域内催化剂表现出的介尺度性质,提升电催化剂的导电性、稳定性和催化活性。因此,关注极端情况之间的介尺度区域,研究各类催化剂在结构、组分、配体、相界面等转换过程中介尺度区域内的形成机理,及其在催化、吸附以及电荷转移过程中的介尺度性质,深入理解各因素调变催化剂性能的介尺度现象,是高效设计和优化催化剂的策略之一,也为筛选制备新型催化剂提供了新的思路和研究方向,对进一步提升催化剂的性能具有重要指导意义。
