富锂锰基三元材料Li1.167Ni0.167Co0.167Mn0.5O2中的氧空位形成*
2022-09-14史晓红陈京金曹昕睿吴顺情朱梓忠
史晓红 陈京金 曹昕睿 吴顺情 朱梓忠
(厦门大学物理科学与技术学院,厦门 361005)
基于密度泛函理论的第一性原理方法,计算了锂离子电池富锂锰基三元正极材料Li1.167Ni0.167Co0.167 Mn0.5O2 中的氧空位形成,讨论了环境温度、压强以及点缺陷的存在对氧空位形成能的影响,还讨论了氧空位对材料容量的影响.结果表明,氧空位的形成能随温度的升高而下降,随氧分压的降低而降低.对于带电氧空位(,空位形成能随着费米能级的升高而增加.研究还表明,氧空位的形成对Li1.167Ni0.167Co0.167 Mn0.5O2 材料中电荷密度分布的影响是相当局域的, 氧空位形成后仅在氧空位附近的Mn 离子周围出现明显的电荷密度的重新分布.此外,计算了氧空位附近存在阳离子空位以及替位点缺陷对氧空位形成能的影响.结果显示,Mn 空位的存在能够明显地促进氧空位的产生.另外,当Mn 被Mo 或Fe 原子替位时,氧空位的产生会受到抑制.
1 引言
锂离子电池因具有工作电压高、能量密度大、环境友好且使用寿命长等优势而得到广泛应用.随着近年来新能源汽车的飞速发展,急需高能量密度的锂离子电池来满足其对动力和里程的需求.其中,制约锂离子电池容量的主要因素为正极材料[1-3].传统的锂离子电池正极材料,如LiCoO2,LiFePO4等[4-9]实际容量均小于200 mA·h/g,且或多或少面临着循环性能或倍率性能差、安全性低、环境不友好、成本高等问题,还不能满足动力汽车对电池的要求.因此,开发高容量的正极材料是提升锂离子电池容量的有效途径之一.近几年来,富锂锰基层状氧化物有望成为新型实用的正极材料,其结构通式可写成xLi2MnO3·(1—x)LiMO2(M为过渡金属),充放电可逆容量能够达到250 mA·h/g 或以上[10].三元正极材料综合了LiCoO2,LiNiO2以及LiMnO2三种正极材料的优点,其化学性质和物理性能随着这3 种过渡金属元素比例的改变而不同,且可以持续优化.另外,其较低的Co 和Ni 的含量能够有效降低电池的成本,增加环境友好性,是很有潜力的动力电池正极材料之一[11-15].
空位是一种典型的点缺陷,对材料的多种性质都有着非常重要的影响.Qiu 等[16]的工作表明,氧空位在材料中提供了一个有利的离子扩散环境.Zhang 等[17]验证了表面氧空位可以提高材料的结构稳定性以及降低电压衰减.因此,研究材料中氧空位的形成对理解材料的相关特性有着非常重要的意义.富锂锰基正极材料的结构可看成是层状单斜相的Li2MnO3(空间群为C2/m) 和层状六方相的LiMO2(空间群为Rm) 的固溶体,这两种空间群的相中锂层和过渡金属层都是交替排列的.本研究通过基于密度泛函理论的第一性原理方法计算了空间群为C2/m的锂离子电池富锂锰基三元正极材料Li1.167Ni0.167Co0.167Mn0.5O2中氧空位的形成能.研究表明,较低的温度和较高的氧分压会使得氧空位形成能增加,从而会抑制氧空位的形成.同时,通过计算电荷密度和电子态密度,进一步观察了氧空位的形成对材料产生的影响.此外分别研究了氧空位附近出现阳离子空位点缺陷和替位点缺陷对氧空位形成的影响.
2 计算方法
研究基于密度泛函理论,以及平面波基和投影缀加波的方法框架,计算程序包为VASP (Viennaab initiosimulation package)[18].交换关联泛函选取的是广义梯度近似 (generalized gradient approximation,GGA)下的Perdew-Burke-Ernzerhof(PBE)泛函[19].平面波截断动能为550 eV,布里渊区的积分采用了Monkhorst-Pack 特殊k点取样方法[20],k点网格为3 × 4 × 3.计算考虑了电子的自旋极化效应,材料的晶格常数和原子位置都进行了充分的优化.为了考虑Li1.167Ni0.167Co0.167Mn0.5O2材料中过渡金属d-电子具有局域性的特点,我们采用了GGA+U方法.参考相关文献[21-24],过渡金属Ni,Co,Mn,Mo,Fe 分别取U=6.0,5.0,5.0,3.5,4.0 eV.三元材料的超原胞含有96 个原子,从超原胞中取出1 个氧原子形成空位时,氧空位的浓度为2%.
3 结果分析与讨论
3.1 Li1.167Ni0.167Co0.167Mn0.5O2 的晶体结构
本文所计算的三元材料Li1.167Ni0.167Co0.167Mn0.5O2的组分可以写为 Li7/6Ni1/6Co1/6Mn1/2O2,其6 倍分子式是Li7NiCoMn3O12(这个组分理论上可以严格计算),而Li2MnO3材料的4 倍分子式是Li8Mn4O12.所 以,可以通过在Li8Mn4O12中 将1 个Li 替换为Ni (或Co),再将1 个Mn 替换为1 个Co (或Ni),从而可以获得所期望的Li7NiCoMn3O12.我们的建模过程是: 为了保持结构是C2/m空间群,从原始空间群为C2/m的Li2MnO3[10]材料出发,首先将过渡金属层中的2 种Li 元素分别用Ni/Co 替换,再分别计算体系的结合能.之后选取其中能量较低的结构,将过渡金属层中的1 个Mn 元素替换成Co/Ni 元素,计算和比较这些结构的结合能,最后根据能量最低的原则获得如图1所示的具有空间群为C2/m的结构模型.由于所有结构在计算时都是经过充分弛豫优化的,所以都是稳定的结构.虽然这样的建模并不能完整地考虑所有结构的可能性,但属于一种合理的可计算的方式.
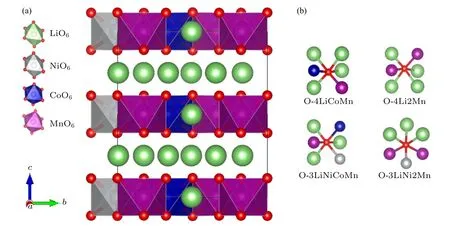
图1 (a) Li1.167Ni0.167Co0.167Mn0.5O2 的晶体结构(其中NiO6,CoO6 和MnO6 八面体分别标识为灰色、蓝色和紫色,氧原子为红色,锂离子为绿色);(b) 氧与周围配位原子的4 种示意图Fig.1.(a) Crystal structure of Li1.167Ni0.167Co0.167Mn0.5O2 (NiO6,CoO6 and MnO6 octahedra are marked by gray,blue and purple,respectively;oxygen and lithium ions are denoted by red and green balls,respectively);(b) diagram of the four coordination pattern of oxygen and surrounding atoms.
由图1(a)可见,所有Li 离子和过渡金属离子(TM=Ni,Co,Mn)都处在密堆积氧晶格的八面体的中心位置,形成LiO6八面体和TMO6八面体.理论晶格常数a=5.0 Å,b=8.70 Å,c=5.07Å(1 Å=10—10m),键角α=90.00°,β=109.19°,γ=90.00° (氧空位计算时的超原胞为2a× 1b× 2c).这些晶格常数与Nakamura[25]等研究的空间群为C2/m的Li1.2Ni0.13Co0.13Mn0.54O2材料的实验值是非常接近的.在富锂锰基材料Li1.167Ni0.167Co0.167Mn0.5O2中,氧存在4 种不同的配位环境(见图1(b)),具体为:
1) O 的周围连接4 个Li,以及1 个Co 和1 个Mn,简写为O-4LiCoMn;
2) O 的周围连接4 个Li 和2 个Mn,简写为O-4Li2Mn;
3) O 的周围连接3 个Li,1 个Ni,1 个Co 和1个Mn,简写为O-3LiNiCoMn;
4) O 的周围连接3 个Li,1 个Ni 和2 个Mn,简写为O-3LiNi2Mn.本文计算了这4 种不同配位情况下的氧空位形成能,将重点讨论空位形成能最小的那个氧空位的形成和物理性质.
3.2 Li1.167Ni0.167Co0.167Mn0.5O2 中单个氧空位的形成能
氧空位形成能的计算对理解材料中氧空位的产生有重要的作用,其大小决定了氧空位在材料中的形成情况.带电荷为q的氧空位形成能Ef()定义为[26,27]
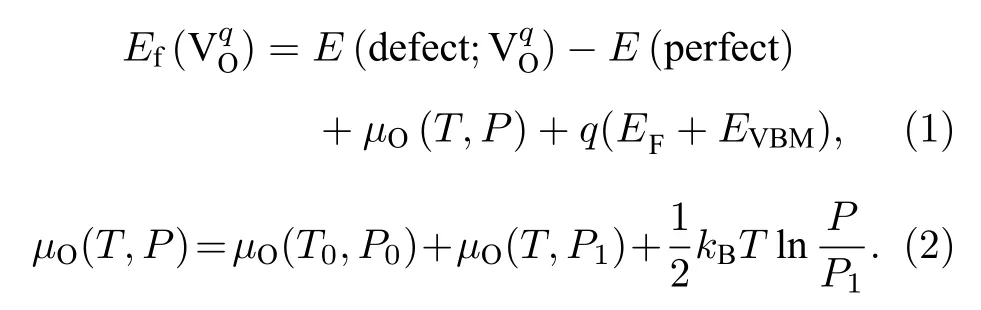
在(1)式中,E(defect;) 表示带有电荷q的氧空位缺陷所对应晶体的能量;E(perfect) 表示没有氧空位的三元材料完整晶体的能量;µO(T,P)是1 个氧原子在给定温度T和氧分压P下的化学势,EF是三元材料完整晶体中的费米能级,其取值范围为0—Egap,Egap是三元材料完整晶体的带隙宽度;EVBM表示价带顶,在这里被取为能量参考点(即EVBM=0).由于是氧的空位,所以考虑了3 种电荷态q,具体为q=0,+1,+2.(2)式中,µO(T0,P0)是1 个氧原子在基态(T0=0 K,P0=0 atm (1 atm=1.013 × 105Pa))时的化学势,可以通过计算O2总能量的一半来得到;µO(T,P1) 表示在特定压强P1(P1=1 atm)下温度对氧化学势的贡献,可以通过相关文献[28]得到;最后一项表示氧分压对氧化学势的贡献,kB为玻尔兹曼常数.
图2 是三元材料中Ni-3d,Co-3d,Mn-3d 和O-2p 轨道的分态密度(partial density of states,PDOS)图.可以看出,计算得到的三元材料是一个磁性半导体,其带隙宽度Egap约为1.10 eV.我们研究了4 种配位情况下的氧空位在不同电荷态、费米能级以及不同外部环境下(温度和压强)的空位形成能.考虑到是氧的空位,所以空位的电荷态只能为q=0,+1,+2,分别记为计算结果列于表1,在配位环境O-4Li2Mn 结构中脱掉其中的O 而形成的空位,其3 种带电态,的氧空位形成能都最低,说明配位环境为O-4Li2Mn 的氧离子最容易形成氧的空位.因此,后文只讨论O 配位环境为O-4Li2Mn 的氧空位形成.
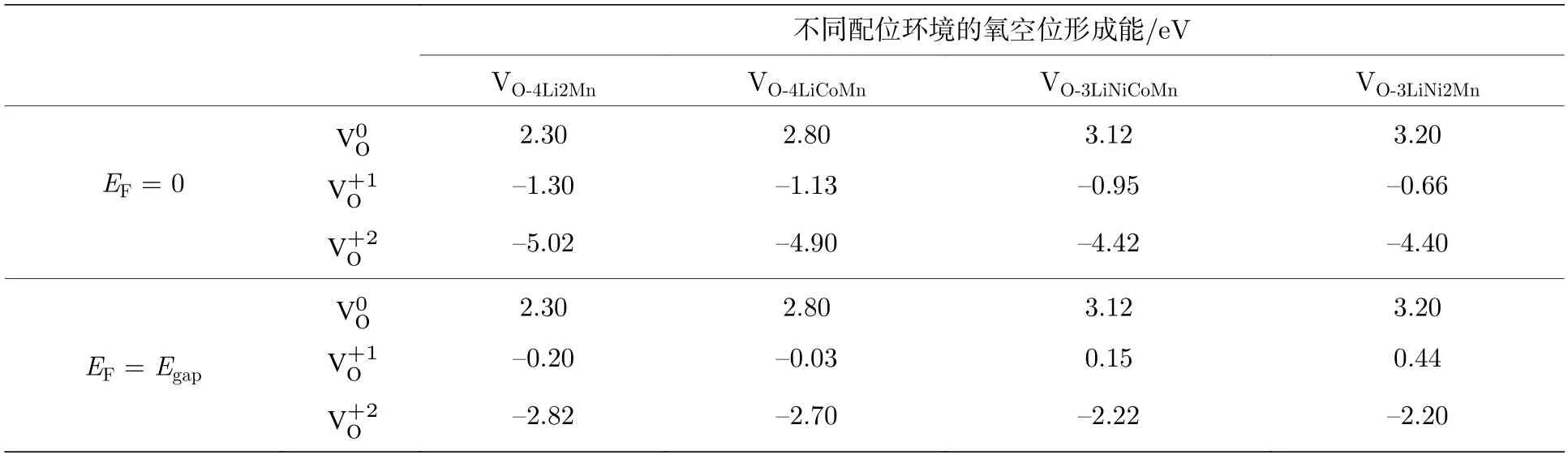
表1 Li1.167Ni0.167Co0.167Mn0.5O2 材料中不同费米能级的3 种带电荷态氧空位的形成能Table 1. The calculated formation energies of non-equivalent oxygen vacancies at different charge states in the bulk Li1.167Ni0.167Co0.167Mn0.5O2 at different Fermi level.
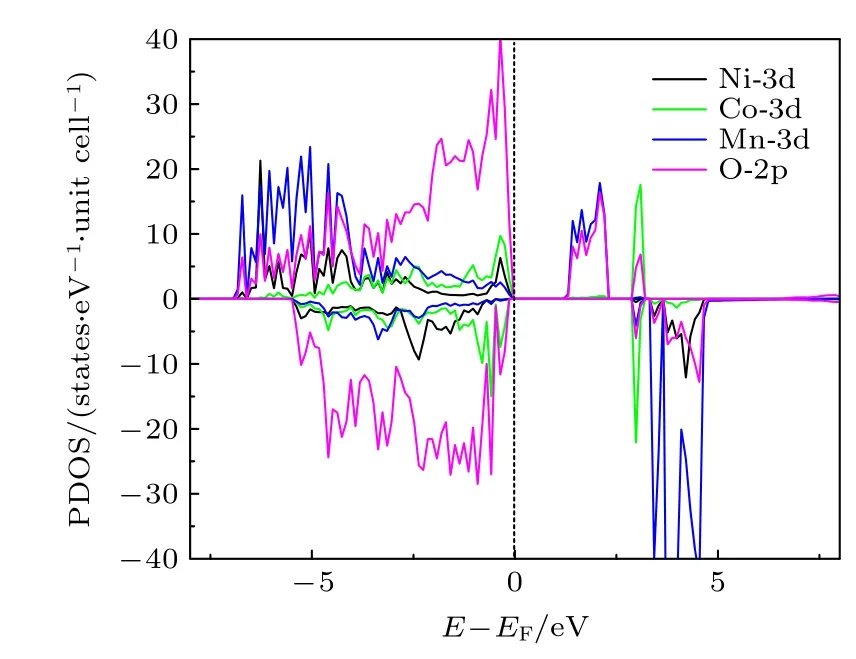
图2 Li1.167Ni0.167Co0.167Mn0.5O2 材料的PDOS 图,虚线表示EF=0Fig.2.PDOS for Li1.167Ni0.167Co0.167Mn0.5O2 material.The dotted line represents EF=0.
环境温度和压强对氧空位的形成有较大的影响,根据(1)式可知,氧空位的形成能是关于氧化学势、费米能级以及电荷态q的函数,而氧化学势又取决于温度、压强等环境条件,即氧化学势由(2)式确定.图3 给出了3 种带电荷态氧空位形成能随费米能级EF(取值范围为 0—Egap) 的变化,其中氧分压P=0.2 bar (1 bar=1.0 × 105Pa),温度变化为T=0—1000 K,间隔为100 K.图3 结果表明,两种带电态氧空位 V的 形成能均随费米能级的升高而增大(中性氧空位形成能与EF无关),同时所有电荷态的空位形成能则随着温度的升高而降低,表明EF的增加不利于氧空位的形成,而温度的升高有利于氧空位的形成.
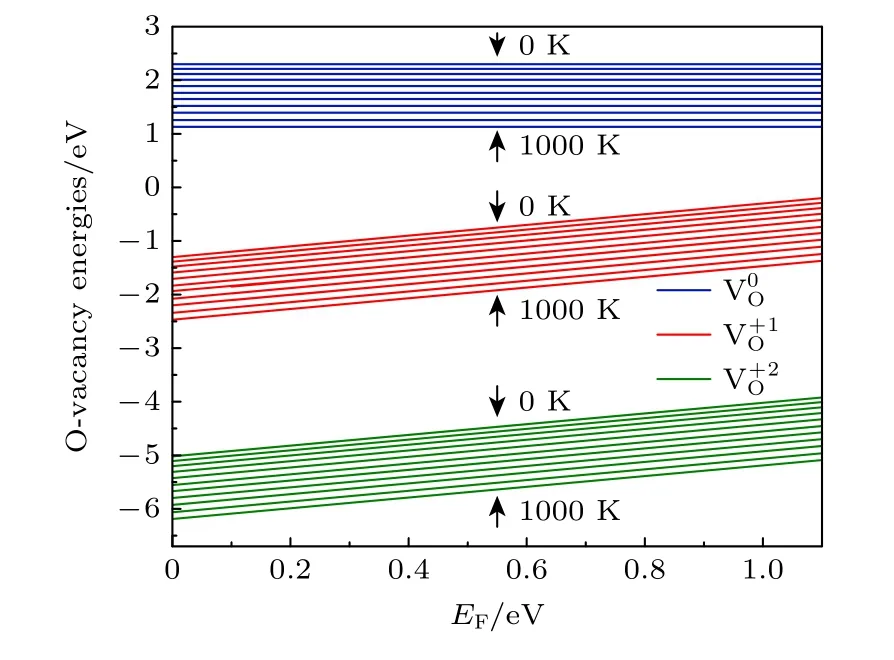
图3 T=0—1000 K,不同带电氧空位的形成能随EF的变化Fig.3.Formation energies of oxygen vacancies in different charge states as a function of E F at T=0—1000 K.
分别取EF=0 和EF=Egap,3 种带 电荷态氧空位的形成能随温度的变化如图4所示.结果表明,当P=0.2 bar 时,不同费米能级(EF=0 和EF=1.10 eV)的 3 种带电荷态 氧空位的形成能有显著差异,电荷态越大,其差异越明显.图4(a)显示,在T=700 K 左右不带电的氧空位形成能为1.88 eV,这与Nakamura 等[25]研究中Li1.2Ni0.13Co0.13Mn0.54O2材料氧空位形成能的实验值非常接近(约为1.92 eV).
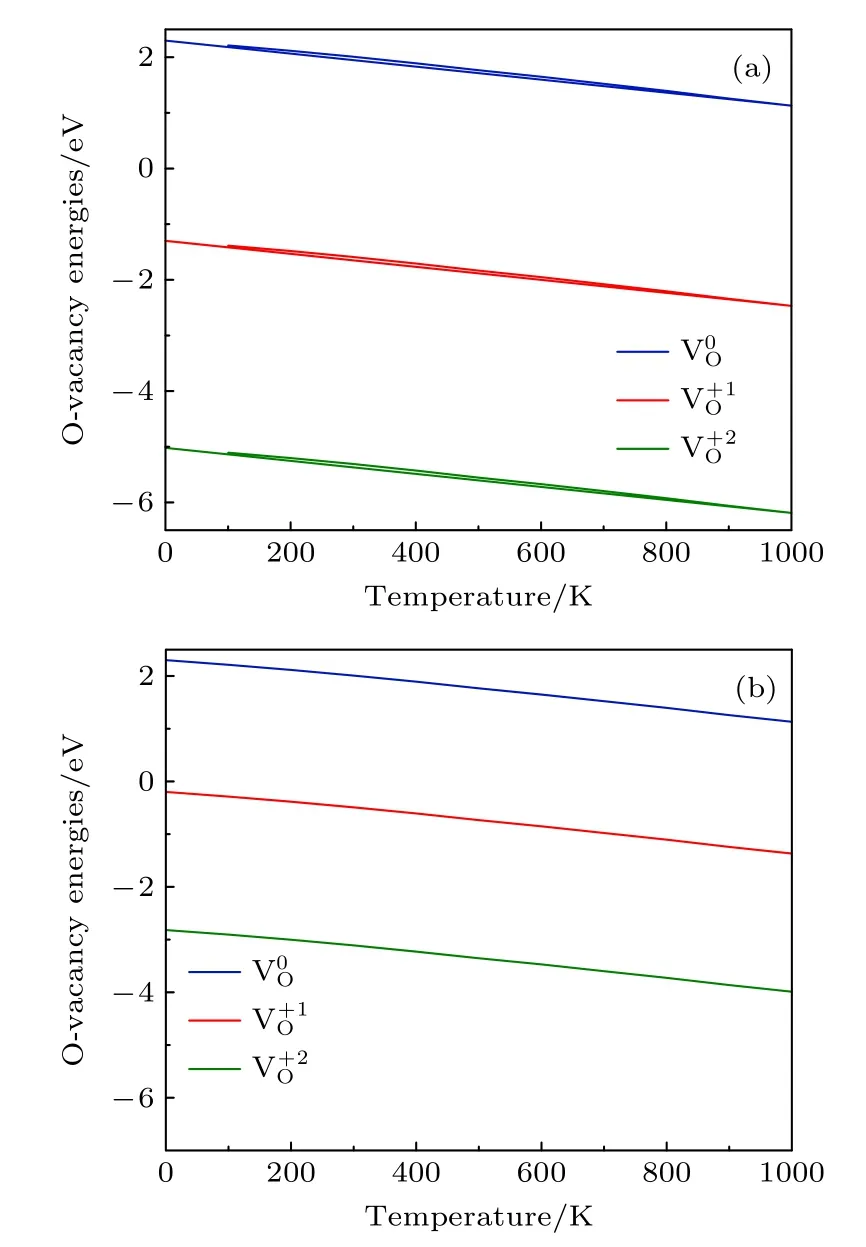
图4 P=0.2 bar,费米能级不同时的氧空位形成能随温度的变化 (a) E F=0 ;(b) E F=Egap .Fig.4.Formation energies of an oxygen vacancy with different Fermi level as a function of temperature at P=0.2 bar: (a) E F=0 ;(b) E F=Egap .
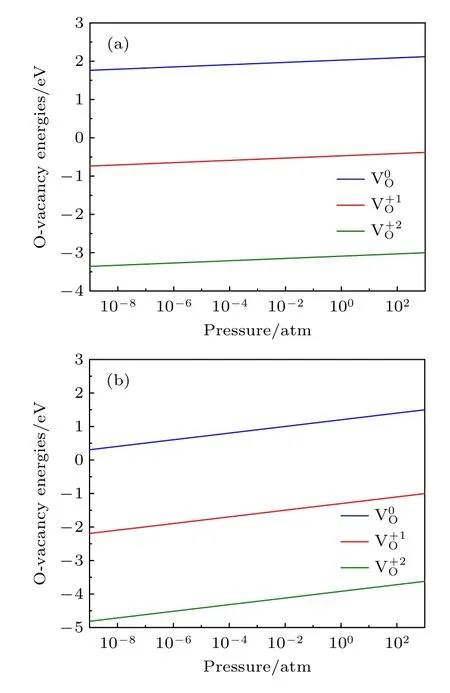
图5 E F=Egap=1.10 eV,温度不同时氧空位形成能随氧分压的变化 (a) T=300 K;(b) T=1000 KFig.5.Formation energies of an oxygen vacancy as a function of oxygen partial pressure at different temperatures with E F=Egap=1.10 eV : (a) T=300 K;(b) T=1000 K.
进一步讨论温度和氧分压同时变化对Li1.167Ni0.167Co0.167Mn0.5O2材料中氧空位形成能的影响(只讨论中性空位的情况),结果如图6 所示.氧空位形成能随温度的升高和氧分压的降低而减小,这与前面的讨论一致.从图6 中颜色变化的梯度可以看出,氧空位的形成能随温度的变化更为剧烈,而随氧分压的变化改变得较不明显.这些计算结果表明,降低温度和增加氧分压可以抑制氧空位的形成,有助于理解氧空位形成的影响因素,为降低锂离子电池正极材料中氧的释放和提高晶格氧的稳定性提供指导.
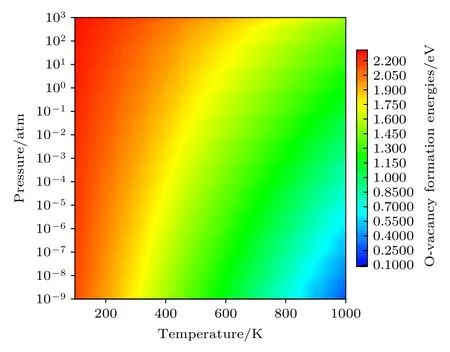
图6 E F=0,中性氧空位 的形成能随温度和氧分压的变化Fig.6.Formation energies of a neutral oxygen vacancyas a function of both the temperature and oxygen partial pressure with E F=0 .
3.3 三元材料中氧空位的电子结构
缺陷的存在可能对材料的性能产生很大的影响,尤其是对电子结构方面的影响.差分电荷密度图可以很好地帮助我们了解Li1.167Ni0.167Co0.167Mn0.5O2材料中原子之间的成键特征以及缺陷对原子间键合性质的影响.差分电荷密度是指同一系统中总电荷密度与单个原子电荷密度的叠加之间的差,即,其中等号右边第一项表示体系的自洽总电荷密度,第二项表示各原子电荷密度的叠加,Rµ是原子位置[29].
第3.2 节指出氧空位形成能最低时的配位情况是O-4Li2Mn,即氧空位周围有4 个锂离子和2 个锰离子,分别将这2 个锰离子标记为Mn-A 和Mn-B.图7(a),(b)分别给出了Li1.167Ni0.167Co0.167Mn0.5O2材料中氧离子及其附近的过渡金属锰离子在没有氧空位和有氧空位情况下的二维差分电荷密度.差分电荷密度的取值范围均为—0.78—0.39 e V/Å,等高线 的间隔 为0.0 3 eV/Å.通 过图7(a)可以看出,TM-O 原子之间除了有明显的电荷转移之外,成键还有明显的方向性,说明TM-O之间的键合作用是明显的离子性和共价性的混合.图7(c),(d)分别为三元材料中氧离子及其附近的过渡金属锰离子在无氧空位和有氧空位情况下的三维差分电荷密度.比较图7(a),(c) 和图7(b),(d)还可以看出,形成氧空位(位置见图7(b),(d))后,氧空位对周围电荷密度分布所产生的影响在这个材料中是非常局域的,仅对氧空位附近的过渡金属Mn-A 和Mn-B 上的电荷分布产生了较明显的影响,这与金属铝中的Al 空位有很大的不同,在金属铝中Al 空位对铝金属电荷分布的影响是相当长程的[30].
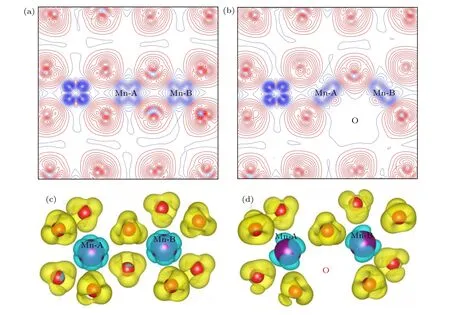
图7 Li1.167Ni0.167Co0.167Mn0.5O2 的二维差分电荷密度 (a) 完整晶体,(b) 有氧空位(红色线表示该区域有电荷聚集,而蓝色线表示该区域有电荷的移出);Li1.167Ni0.167Co0.167Mn0.5O2 的三维差分电荷密度 (c) 完整晶体,(d) 有氧空位Fig.7.2D charge density plots of Li1.167Ni0.167Co0.167Mn0.5O2: (a) Pristine;(b) with oxygen vacancy (The solid and dashed lines represent the accumulation and depletion of charges relative to the independent atoms,respectively);3D charge density plots of Li1.167Ni0.167Co0.167Mn0.5O2: (c) pristine;(d) with oxygen vacancy.
图7 对含有氧空位材料的差分电荷密度的分析显示,氧空位所产生的影响是非常局域的.另外,由于氧空位形成能最低时氧的配位情况是O-4Li2Mn,所以在这里只需讨论氧空位周围的2 个锰离子(即Mn-A 和Mn-B)的电子态密度.图8 给出了氧空位附近的过渡金属Mn-A 3d 和Mn-B 3d 电子的PDOS 图,比较了3 种电荷态下的氧空位在完整无缺陷体系(Perf.Mn)和含空位体系(Def.Mn)中的电子态密度.除了氧空位的Mn-B 3d-电子态密度图(即图8(e))之外,所有电荷态的氧空位都在其附近的Mn-A 和Mn-B 原子3d-态密度图的费米能级上出现了态密度峰,即氧空位的引入导致了带隙中缺陷能级的出现(虽然这些电子态都不多).
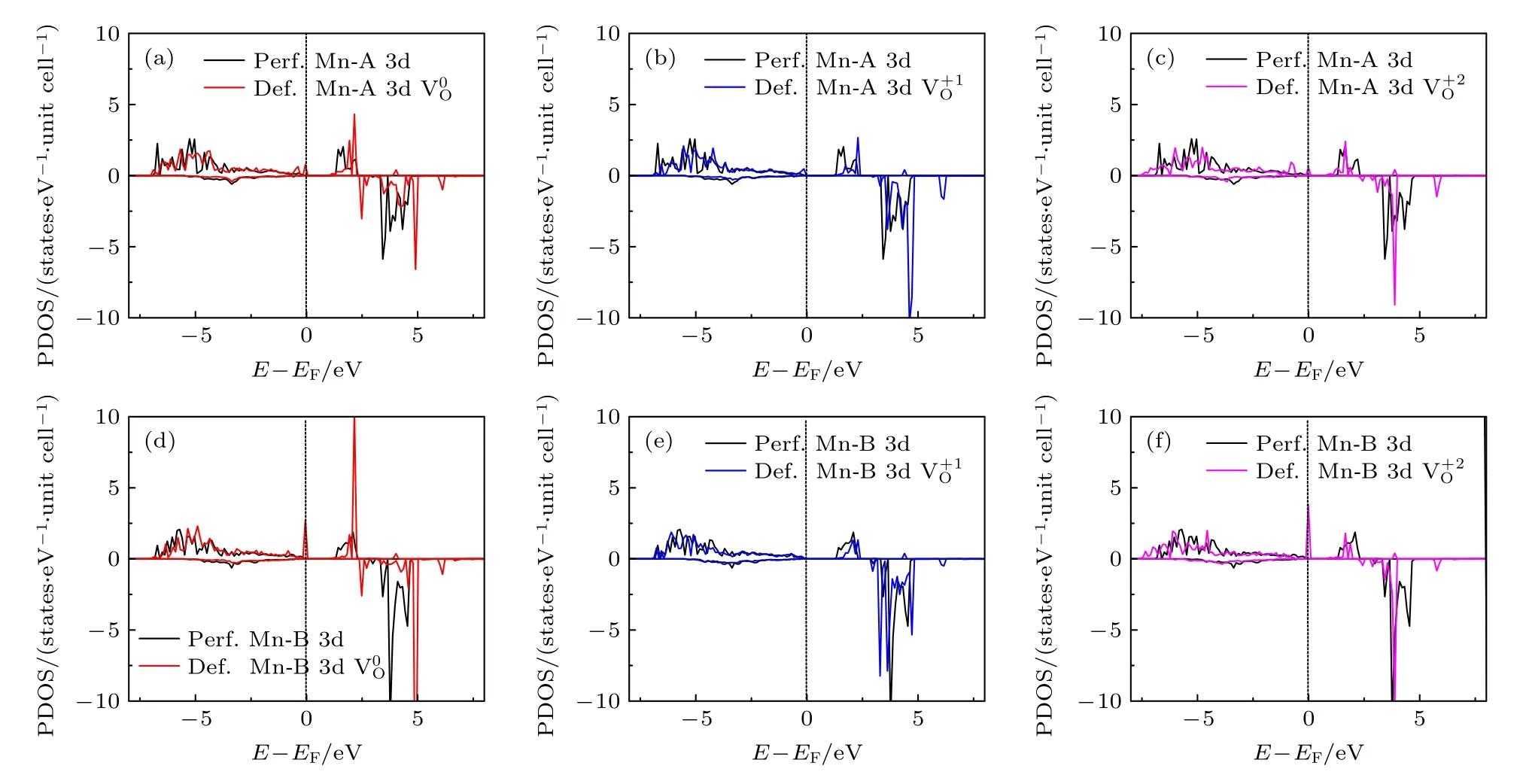
图8 氧空位附近Mn-A 3d (a)—(c)和Mn-B 3d (d)—(f) 电子在不同电荷态下的PDOSFig.8.PDOS of Mn-A 3d (a)—(c) and Mn-B 3d (d)—(f) electrons in different charge states near the oxygen vacancies.
下面讨论氧空位的存在对材料容量的影响,容量的计算公式为

其中x为脱嵌的电荷数.由(3)式可知,在不考虑氧空位的存在对体系结构稳定性的影响时,氧空位存在时材料的总分子量会稍稍减少,可以使材料的容量稍稍增加.实际上,氧空位的存在使得材料中总的负价数减少,总 负价数=(N -δ)×(-2),N为原胞中的氧原子数,δ为原胞中的氧空位数.为了保持体系的电中性,过渡金属的总正价数也会相应地减少,最终导致可以有更多来自过渡金属的电荷补偿,而使得材料的容量稍稍增加.
3.4 点缺陷的存在对氧空位形成的影响
材料中存在氧空位之外的其他点缺陷将对氧空位的形成产生重要的影响.我们研究了氧空位附近存在的其他空位以及替位点缺陷对氧空位形成能的影响.首先计算了氧空位附近存在阳离子Mn 空位 VMn对氧空位 VO形成能的影响.氧空位与近邻空位 VX之 间的相互作用能Eint计算表 达式为[31]
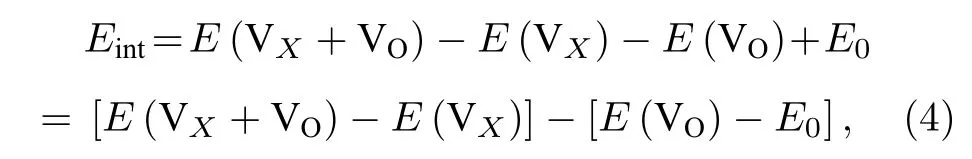
其 中,VX代表氧 空位附近的 VMn阳离子空位.E(VX+VO),E(VX) 和E(VO) 分别表示一个复合空位 VX+VO的能量,单个阳离子空位 VX和单个氧空位的VO的能量,这些都是由同一个晶胞计算出来的.E0是无缺陷的原始材料的能量.Eint< 0表 明 VX和VO相互吸引,否则 VX和VO相互排斥.Eint可以看作是其他空位的存在引起氧空位形成能的变化.图9 给出了氧空位与近邻阳离子空位VMn之间的相互作用能.结果显示,Eint是负值(即阳离子空位对氧空位有吸引作用)将会导致氧空位的形成能降低,表明了氧空位附近存在阳离子Mn 空位缺陷时将有利于形成氧空位.
我们还计算了氧空位附近的Mn 被Fe 和Mo 替位时对氧空位形成能的影响.替位点缺陷与氧空位之间的相互作用能Eint表达式为[31]
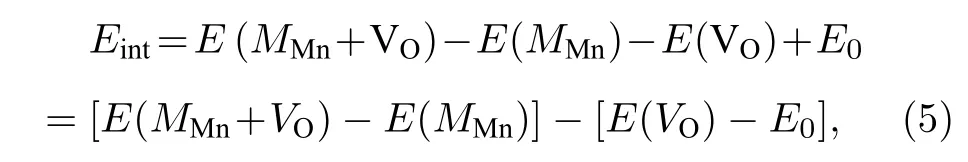
其中MMn代表用1 个M原子 (M=Mo,Fe) 替位氧空位附近的Mn 原子.E(MMn+VO) 和E(MMn)分别表示含有复合缺陷MMn+VO和单个替位点缺陷MMn的能量.图9 展示了2 个替位点缺陷MMn(M=Mo,Fe) 与氧空位的相互作用能.显然,两个相互作用能都是正值的,即氧空位近邻的Mn 被Mo 或Fe 原子替位时都将导致氧空位形成能的增加,所以这种Mn 被替位的情况不利于氧空位的形成,换言之将抑制氧空位的产生.
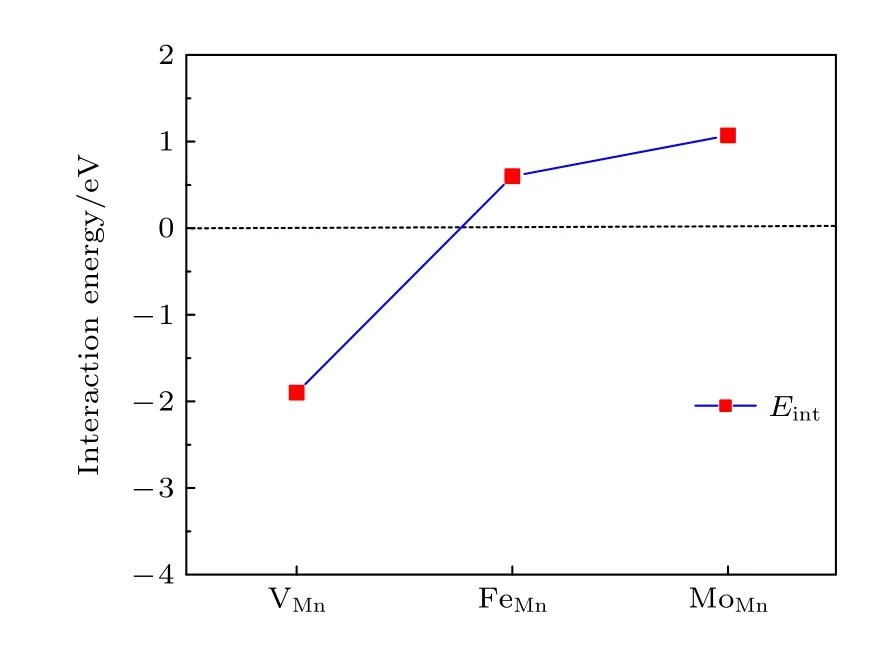
图9 氧空位与其邻近点缺陷的相互作用能,缺陷包括阳离子空位 V Mn 以及氧空位邻近 的Mn 被Fe 和Mo 替位Fig.9.Interaction energies of an oxygen vacancy and its neighboring point defects,including vacancies V Mn and substitutional FeMn,MoMn point defects,respectively.
4 结论
本文基于第一性原理方法计算了锂离子电池富锂锰基三元正极材料Li1.167Ni0.167Co0.167Mn0.5O2中氧空位的形成,研究了氧空位的形成能与环境温度、氧分压以及点缺陷的存在对氧空位形成能的影响.研究发现,温度的升高或氧分压的降低都使得氧空位的形成能减小,即较低的温度和较高的氧分压会使得氧空位形成能增加,从而会抑制氧空位的形成.带电氧空位的形成能还会随费米能级EF的升高而变大.当EF=0 和EF=Egap,压强P=0.2 bar 时,带电氧空位的形成能在0—1000 K 的温度区域内均为负值,说明Li1.167Ni0.167Co0.167Mn0.5O2中会存在低温下少量析氧的现象(当电子和氧被同时“电离”的时候).当氧空位形成后,氧空位对材料电荷密度的影响非常局域,仅在氧空位附近的Mn 离子附近出现明显的电荷重新分布.我们还研究了氧空位附近存在阳离子点缺陷对氧空位形成能的影响.结果表明,当氧空位附近出现Mn 空位时,将有利于形成氧空位.当氧空位近邻的Mn 原子被Mo 或Fe 原子替位时,都导致了氧空位形成能的增加,所以这些Mn 原子被替位的情况将抑制氧空位的产生.这些计算有助于理解该富锂锰基正极材料中氧空位的形成,并为减少材料中氧的释放和提高这种正极材料中晶格氧的稳定性提供指导.
