光电解水产活性氢/氧耦合加氢/氧化过程用水滑石基纳米材料
2020-10-27来天艺王纪康李天白莎郝晓杰赵宇飞段雪
来天艺,王纪康,李天,白莎,郝晓杰,赵宇飞,段雪
(北京化工大学化工资源有效利用国家重点实验室,北京100029)
引 言
随着世界经济的发展与化石能源的日益枯竭,发展清洁可再生能源已成为一项迫切的任务。氢能具有储量丰富、清洁高效、可再生等诸多优势,被普遍认为是21 世纪最具有长远发展前景的无污染绿色清洁能源之一。目前已经在发电、交通以及工业加氢催化等领域得到广泛应用,受到政府、科技界和产业界的密切关注,并已纳入我国能源发展规划,预计在2050年氢气的利用将达到5.39亿吨。目前氢气制备主要通过化石能源重整,即通过水煤气变换过程制氢(CO +H2O= ===== CO2+H2),然而该方法在制备过程中存在化石能源大量消耗和CO2等温室气体大量排放等问题,易对环境造成大规模污染。同时,该合成方式生产成本相对较高,依然需要大量的化石能源消耗,因而未能从根本上达到节约能源的目的,且所得氢气产物通常含有难以去除的CO等杂质,使得该方法有待进一步优化提高[1]。自然界中水资源储量丰富,因而通过光/电催化分解水制氢被认为是有望替代化石能源制氢的一种优越的制备方法,也是目前制氢的唯一清洁、可持续发展的终极制氢路线。通过合理选择调控催化剂材料结构及制备合适的电极材料,再通过光电作用催化分解水过程可制备得到高纯度氢气,不仅节约化石能源,还可实现CO2等温室气体的零排放,真正实现能源可再生,从根本上解决世界能源问题[2]。但是,目前电解水制备氢气仍存在多种问题及困难,其中电解水过程需消耗大量的电力资源是制约电解水制备氢气发展的主要因素。虽然理论上电解水过程的过电位仅为1.23 V,但在实际生产过程中,电解水过程的过电位通常达到1.8~2.3 V,远远高于理论电势,因此极大地限制了通过电解法制备H2在实际工业生产中的应用[3−4]。在光电水分解阴极制氢过程(HER 过 程)中[5−6],往 往 会 忽 略 阳 极 生 成O2过 程(OER),甚至牺牲氧导致成本增加,造成较低产氢效率。OER 过程涉及到四电子转移过程且反应能垒较高[7],使得该过程通常需要较高的过电位同时反应过程动力学较为迟滞,相较于水分解产H2半反应来说更难发生,因而是限制水分解制备氢气的另一个重要因素[8−9]。
通过光电催化方式分解水产生的氢能源,不仅可直接用作燃料供能,亦可用于工业催化加氢过程制备得到高附加值精细化工产品[10]。同时,光电解水产生的活性氧物种,亦可通过耦合工业氧化反应制备苯酚等高附加值精细化学品[11]。通过上述光电耦合反应的方式,不仅可以避免传统催化条件下苛刻的反应条件[12−13],还可以避免氢气、氧气或双氧水等反应原料的过度消耗,从而实现资源节约的目的[14]。此外,通过光/电等外场调控和催化剂表面微结构优化,还可以促进高附加值目标产物生成以及直接向H2O2转化[15]。该过程在很大程度上利用光能电能,使得工业催化加氢及氧化过程均可以在温和的反应条件下进行,在提高水分解制氢气效率的同时,产出高附加值的氧化产物,进而降低产氢成本。此外,该过程通过将氧化半反应的4 电子过程变为2 电子过程,以达到降低氧化活化能、协调传质、降低活性氧与活性氢的复合,最终达到提高产氢效率的目的;而利用电解水产生的活性氢进行耦合加氢过程,为活性氢提供多种反应途径和多种目标产物,水分解制氢不仅限于提供能源,还可以拓展至广阔的绿色加氢/氧化化工过程;并且借助光能,促进电解水效率并提高产物选择性,进一步降低产氢成本,从而提高催化过程转化效率,同时减少了该过程中因储存、转化造成的能量损耗问题,被认为是解决能源问题的一类重要催化反应。
实现上述耦合过程的重点和难点为将两个不同的反应有机结合,使得该两类反应可以在同一体系、相同的催化条件下同时进行。催化该耦合过程既要考虑到催化过程中产生活性氢(*H)/活性氧(*O)的还原/氧化能力,同时又要避免氢气(HER 过程)及氧气(OER 过程)生成的竞争,因而对催化剂的选择和调控是实现该过程的关键[16]。层状双金属复合氢氧化物(又称水滑石,layered double hydroxides,LDHs),结 构 式 为[M2+1−xM3+x(OH)2]x+[Ap−x/p]x−·mH2O,因其具有层板金属元素可调、层间阴离子可控、层板尺寸和间距可变等高度可调性,在光电催化领域得到了广泛应用[17−18]。目前,对于LDHs 及其衍生物应用于光电催化产活性氢/氧方面,已取得系列研究进展[19]。LDHs 表面具有丰富的活性位点、可调控的能带及界面结构、可调变反应过程的能量势垒等,因而在目标反应中可通过LDHs 结构的可控调变,实现水分解过程中活性氢/氧与催化加氢还原/氧化过程反应物有机结合,同时避免*H/*O 结合产生氢气/氧气等副产物[20]。目前,LDHs 在光/电催化分解水产氢气,光电解水耦合CO、CO2、N2分子还原、加氢等反应,以及光电解水耦合苯/苯系物催化氧化等方面均取得突破性研究进展[21]。本文重点介绍近年来LDHs 基材料用于催化光电分解水过程、现代化学工业中常用的催化加氢还原/氧化过程,光电解水产*H 及*O 中间体耦合加氢还原/氧化反应的最新进展,并指出了该领域目前尚存在的问题。
1 LDHs应用于光/电催化分解水
LDHs 作为一类典型的二维层状氢氧化物材料[图1(a)],其层板元素组成、比例、层间阴离子种类、粒径、厚度均具备高度可调性,可通过多种手段调控催化剂的能带结构。理论计算结果表明,LDHs材料可以实现还原电位在−0.5~4 V范围内的广泛调变[图1(b)],如此大范围的控制,为后续分解水耦合还原过程提供了可能;其氧化电位亦具有大范围可调控的特点[22−23]。进一步通过调控LDHs 材料独特的层板厚度和尺寸,在光电催化分解水过程中可以明显促进载流子分离传输,显著提升其催化转化效率,被广泛用于光/电催化分解水过程。通过调变LDHs 材料元素组成及缺陷结构,对LDHs 材料的能带结构进行进一步合理调控,可以实现高效光电催化分解水的目的[24]。
1.1 光电分解水过程机理
碱性环境下水分解过程的析氢及析氧反应均涉及活性中间体*H及*O的生成及消耗。
水分解过程为:
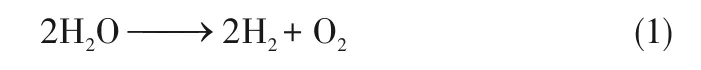
碱性条件下的析氢半反应为:

推测碱性条件下析氢过程反应机理为:

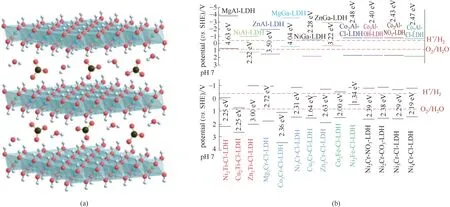
图1 LDHs材料结构示意图(a)[17];常见LDHs材料pH=7时的价导带位置图(b)[22]Fig.1 Schematic illustration of LDHs(a)[17];Conduction band and valence band potentials of some common LDH materials versus NHE at pH 7(b)[22]
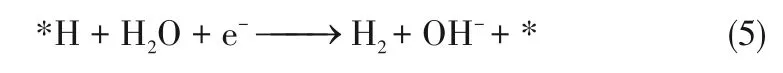
碱性条件下的析氧半反应为:

推测碱性条件下的析氧过程反应机理为:
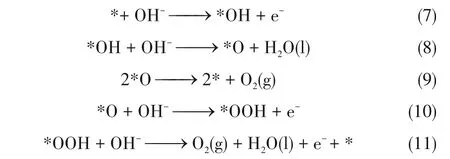
综上[式(1)~式(11)],碱性条件下,水分解析氢过程中,水分子与催化剂活性位点相结合产生*H中间体,该活性中间体两两结合生成H2分子。而析氧过程中,碱性溶液中的OH−与活性位点相结合形成活性OH(*OH)中间体后结合OH−脱去H2O 后形成*O,*O进一步结合形成O2分子[25]。
1.2 光催化分解水

图2 ZnTi−LDH高分散结构示意图(a);Ti基LDH光催化分解水产氢性能对比(b);甲醇溶剂中·ESR检测图谱(c)[29];Ti3+自掺杂的NiTi−LDH能带结构示意图及可见光照条件下O2生成过程示意图(d);不同厚度NiTi−LDH荧光光谱(e);以10−2 mol·L−1 AgNO3 为牺牲剂不同厚度NiTi−LDH的分解水产O2性能示意图(f)[30]Fig.2 A polyhedral representation of the ZnTi−LDH structure(a);H2 evolution productivity of MTi−LDH(b);ESR spectra recorded for DMPO−·O2−in methanol dispersion(c)[29];Proposed structural model of energy states for the Ti3+self−doped NiTi−LDH and schematic illustration of the O2 evolution process over the NiTi−LDH nanosheet under visible−light irradiation (d);Fluorescence spectra(e);O2 evolution from aqueous solution using 10−2 mol·L−1 AgNO3 as the sacrificial acceptor under visible−light using NiTi−LDH with different thickness(f)[30]
光能直接转化为化学能是目前能源领域的研究热点之一,其中光解水制备氢气是一种极具潜力的技术,该反应中催化剂的使用可大大提高光催化分解水的效率。设计性能优异的光解水催化剂的关键在于对催化剂表界面结构的合理调控,进而获得较好的电子空穴分离效果,实现光解水的高效催化转换。其中TiO2在光催化分解水领域一直是研究热点材料。研究表明,过渡金属阳离子高分散的网状结构可以显著提高光生电子空穴的迁移效率,从而进一步提高光解水的转换效率[26]。自1972 年Honda 等[27]发现可利用TiO2作为催化剂实现光电催化分解水以来,多种纳米材料显示了优越的光催化分解水性能;García等[28]首次报道了Zn基LDHs材料可以在可见光条件下分解水产氧气,在λ>410 nm时,量子效率高达60.9%。结合LDHs层板金属离子的高分散特性以及LDHs 正八面体的结构优势,Duan 等[29]合 成NiTi−LDH、ZnTi−LDH 等 多 种Ti 基LDHs,实现了TiO6正八面体结构在催化剂中高度分散的效果[图2(a)]。其中ZnTi−LDH 催化剂的光催化产氢效率可达31.4 μmol·h−1,为常规层状高分散Ti基催化材料K2Ti4O9产氢效率的18倍,实现了Ti基催化剂光催化性能的显著提升[图2(b)]。通过瞬态表征证实材料在催化水分解过程中存在活性氧中间体[图2(c)],为该过程进一步耦合现代工业催化加氢及氧化过程奠定了基础。为进一步提升Ti 基LDHs材料催化性能,Duan 等[30]着眼于材料尺寸的调控。采用反向微乳法,通过改变表面活性剂与水的比例对NiTi−LDH 尺寸形貌进行调控,成功合成横向尺寸为30~60 nm,厚度约为2 nm 的超薄NiTi−LDH。该LDHs 材料中存在大量的Ti3+缺陷和氧缺陷,经理论计算(DFT)证实,Ti3+的存在显著降低了催化剂的导带位置[图2(d)],根据荧光光谱表征结果显示,超薄的Ti 基LDHs 具有更好的光生电子与空穴分离的能力,建立了纳米材料微观尺寸与材料催化性能的有机联系[图2(e)]。其中层板尺寸最小(约30 nm)的NiTi−LDH−1 催化材料实现了最高2148 mmol·g−1·h−1的产氧效率[图2(f)],相较于前人的工作,催化性能有了进一步的提升。为提高光生电子空穴的分离效率,Ti 基LDHs 材料也常常与石墨烯等材料结合得到复合型催化剂材料,在光催化分解水领域应用广泛[31]。
除Ti 基LDH 之外,ZnCr−LDH 也被广泛应用于光催化分解水产氧过程,但因ZnCr−LDH 材料在催化过程中生成的光生电子空穴对极易复合,因而利用ZnCr−LDH 进行催化水分解时,需要对其电子结构进行改良,与其他半导体材料结合形成异质结用于催化水分解过程是目前常用的策略[32]。Wei 等[33]采用P 型半导体材料Cu2O 与ZnCr−LDH 复合形成Z型异质结,制备得到了Cu2O@ZnCr−LDH 核壳结构[图3(a)],并在ZnCr−LDH 层板间插入S2O2−3对该异质结构进行进一步修饰。通过该方式负载制得的Z型异质结结构催化剂[图3(b)],ZnCr−LDH 导带中产生的光生电子可迁移至Cu2O 价带中,有效促进电子空穴对的分离及传导过程,进而实现对水分解过程效率的提升[图3(c)]。在此基础上,该课题组通过共轭π 键在异质结结构间形成供体−桥式连接−受体特殊的传输结构,进一步提高了催化剂中的电子传导效率[图3(d)],将全解水产氢气和氧气的性能整体提高4倍,实现了性能的突破性进展,并通过多普勒拓展正电子湮灭光谱技术(CDB−PAS),实现了对光催化分解水过程产生的光生电子反应过程首次追踪,更确切地揭示了光催化过程的构效关系[图3(e)],为异质结结构催化剂在高效光分解水方面的应用提供了思路[34]。
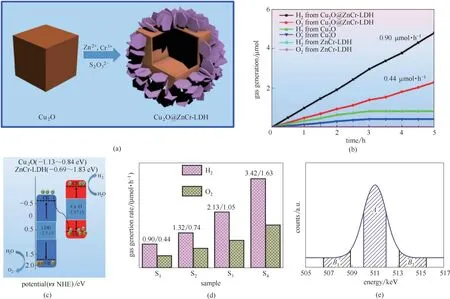
图3 Cu2O@ZnCr−LDH制备过程及核壳结构示意图(a);Cu2O@ZnCr−LDH光催化水分解气体产率随时间变化(b);Cu2O@ZnCr−LDH体系中光生电子分离传输方式示意图(c)[33];Cu2O@ZnCr−LDH产气性能对比(d);Cu2O@ZnCr−LDH CDB−PAS 表征中S及W参数意义(e)[34]Fig.3 Schematic illustration for the preparation of Cu2O@ZnCr−LDH hollow coreshell photocatalyst(a);Rate of gas generation as function of irradiation time(b);Schematic illustration for the photoexcited electron separation/transport in the Cu2O@ZnCr−LDH system(c)[33];Gas generation rate(d);Schematic definition for S−and W−parameters in CDB−PAS measurements of Cu2O@ZnCr−LDH(e)[34]
1.3 电催化分解水
LDHs 层板结构中的不饱和位点在电催化反应过程中被认为可作为催化过程的活性位点,且LDHs 材料因层板尺寸可调及层板中缺陷的可控等特性,被广泛应用于电催化分解水过程。在催化剂的选择中,催化剂的本征活性及合成过程中如何巧妙地引入活性位点是实现LDHs 高效催化电解水性能的决定性因素,其中NiFe−LDH、CoFe−LDH 等多种LDHs 因其微观电子结构的特殊性,被广泛认为是电催化分解水制氧过程最有潜力的催化剂。在OER 过程中,提升其催化性能的常用方法包括在LDHs 主体层板中引入缺陷作为活性位点,从而合理调整反应中间产物的能量势垒,以达到降低催化过程过电位的目的[35−36]。Du等[37]提出可通过负载的纳米团簇与LDHs 材料形成的界面结构,为OER 过程活性中间体的三维吸附提供合适的空间位点,因而实现了OER 性能的显著提升。Zhang 等[38]通过贵金属单原子负载方式,显著降低OER 过程*O 中间体的能量势垒,可显著提升OER 过程的催化性能。Sun等[39]使用2 mol·L−1NaBH4溶液对NiFe−LDH 进行浸泡还原处理[图4(a)],在NiFe−LDH 中引入丰富的氧缺陷作为OER 过程的活性位点,在10 mA·cm−2电流密度条件下,过电位仅为200 mV [图4(b)]。由DFT 计算可知,含氧缺陷的NiFe−LDH 可有效降低OER反应过程中生成*O中间体的反应能垒,进而使该反应可在更低的外加能量条件下进行[图4(c)]。除还原处理方式外,对LDHs 材料进行剥离亦是在材料中引入缺陷提高电催化性能的一种有效途径。Wang 等[9,40]采用等离子共振效应,通过Ar 气等离子体对CoFe−LDH 进行电离处理[图4(d)],实现了对LDHs 的干法剥离,证实其在该剥离过程中引入大量的氧缺陷和金属缺陷[图4(e)、(f)],显著提高了CoFe−LDH 的催化性能。Zhang 等[41]通过超声剥离法在LDH 层板中引入大量缺陷,制备横向尺寸小于3 nm 的单层超小NiFe−LDH,提高LDHs 层板表面的不饱和位点数目,有效降低了LDHs OER 过程的过电位,提高了能量转化效率。
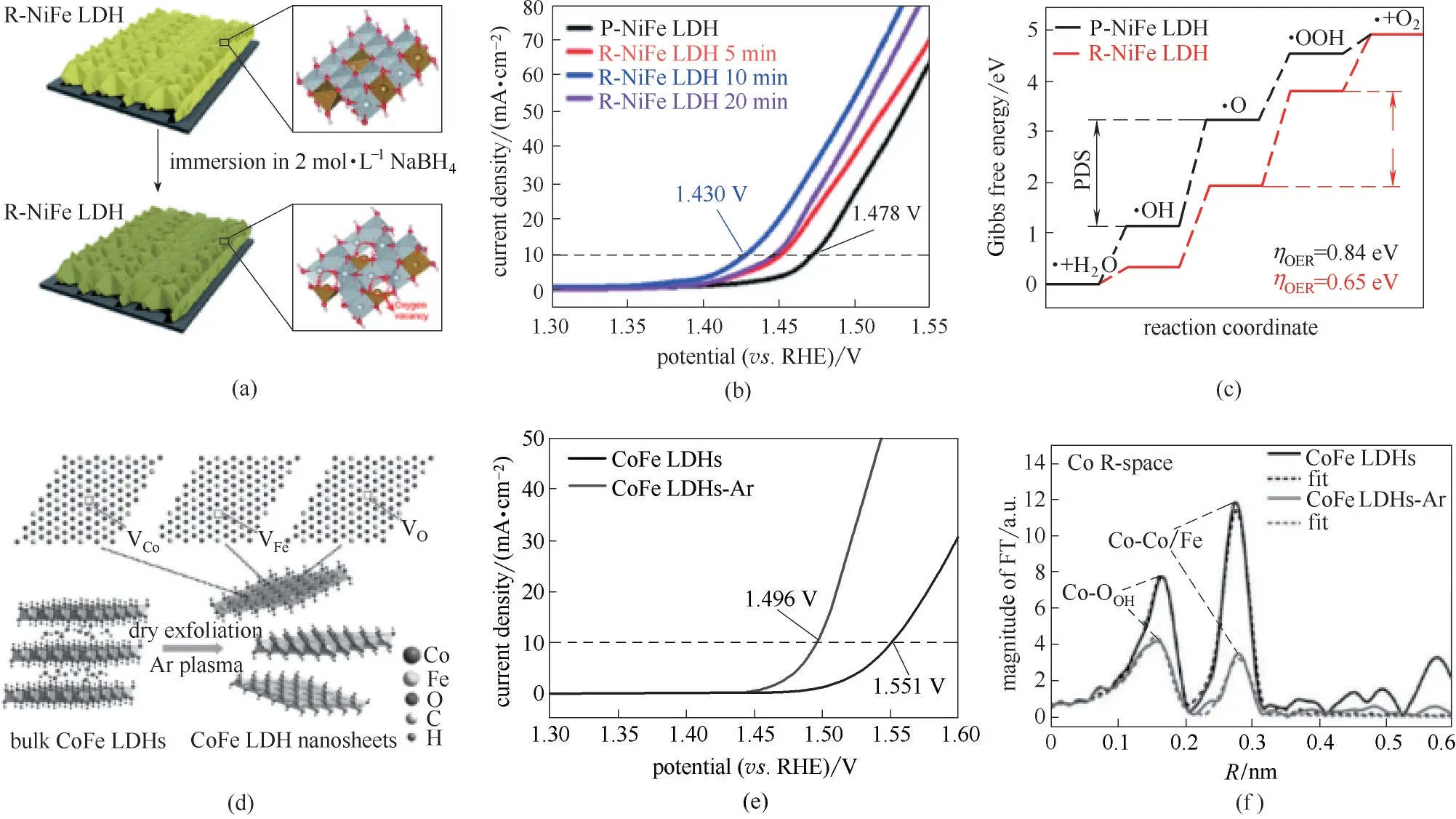
图4 NiFe−LDH 纳米棒电极中引入O缺陷过程示意图(a);NaBH4溶液浸泡处理NiFe−LDH LSV曲线(b);NaBH4溶液浸泡处理NiFe−LDH DFT计算结果(c)[39];Plasma过程对CoFe−LDH进行干法剥离示意图(d);块体CoFe−LDH及超薄CoFe−LDH LSV 性能(e);Plasma过程处理后的CoFe−LDH Co元素XAFS R空间测试结果(f)[9]Fig.4 Schematic illustration of introducing oxygen vacancy defects to NiFe−LDH nanoarray electrode (a);Linear sweep voltammetry polarization curves of as−prepared NiFe−LDH after NaBH4 treatment(b);Free energy plots calculated results of OER process NiFe−LDHs treated by NaBH4(c)[39];CoFe−LDH nanosheets by Ar plasma exfoliation(d);LSV curves of bulk CoFe−LDHs and ultrathin CoFe−LDH nanosheets(e);Magnitude of the k3−weighted Fourier transforms of the Fe edge XANES spectra for bulk CoFe−LDH and ultrathin CoFe−LDH(f)[9]
LDHs 材料同样广泛应用于电催化制氢过程,因HER 过程通常倾向于在酸性环境下进行,而OER过程通常需要碱性的电解质条件[42−43]。LDHs 材料作为一种典型的碱性氢氧化物材料,在酸性介质中循环稳定性不尽如人意,对LDHs 结构进一步处理,是实现LDHs 基材料全水解的关键。Shao 等[44]以CoNi−LDH 和NiFe−LDH 为前体,通过磷化过程和电化学沉积过程,成功合成CoNiP@LDH 核壳结构[图5(a)],通过将该材料负载在电极片表面后进行电解水过程[图5(b)],证实该电极材料可实现长达500 h 以上的全水解循环稳定性,在10 mA·cm−2电流密度条件下,过电位仅为210 mV[图5(c)]。Shao 等认为在该结构中,过渡金属磷化物CoNiP 中的金属位点为催化HER 反应的主要活性位点,且根据X 射线光电子能谱分析(XPS)表征结论,Fe 元素的价态显著升高,高价的Fe元素被认为是提升OER过程的关键因素,两种因素协同作用使得合成的CoNiP@LDH 全水解性能显著提升。除对LDHs 材料进行P 化处理提高其HER 催化效率外,将LDHs 材料进行N 化、Se化及B 化处理也有广泛报道。Zhang 等[45]以NiFe−LDH 为前体,通过原位生长结合热氨分解法制备得到NiFeN 材料用于HER 催化过程。NiCo−LDH 通过Se 粉与NaBH4溶液混合还原处理[46]以及B 元素掺杂[47]均可显著提升HER 性能。Ostrikov 等[48]通过N及P 原子对NiVFe−LDH 材料的掺杂,HER 催化过程过电位仅为79 mV,同时其OER 催化过程过电位仅为229 mV。同时,通过DFT 计算结果可知[图5(d)],经过N 及P 原子掺杂的LDH 材料中形成*H 过程的Gibbs 自由能更低,证明HER 过电位降低的根本原因是降低了*H的能量势垒,促进了H2的生成。
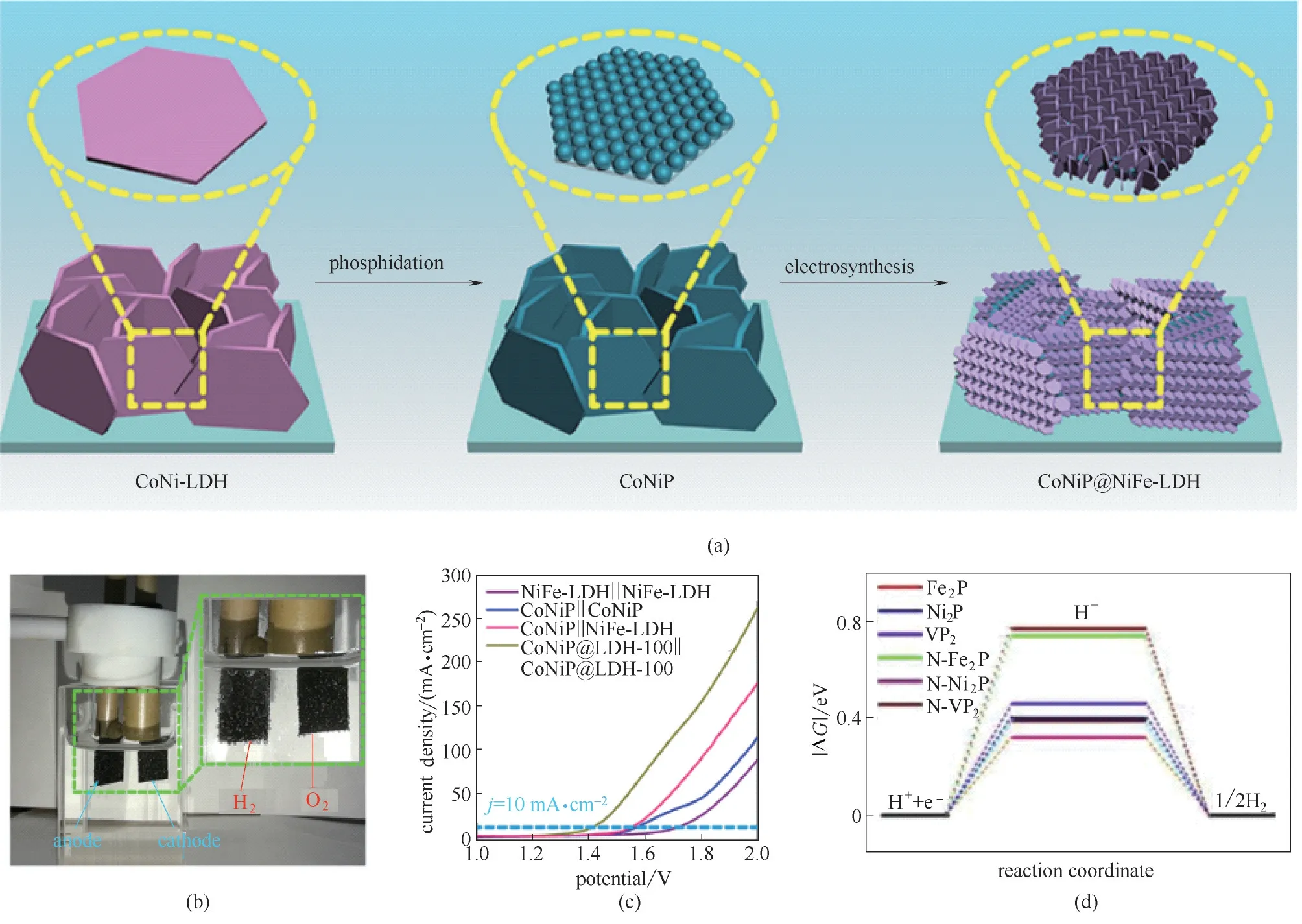
图5 CoNiP@LDH层状阵列制备过程示意图(a);水分解体系实物图(b);不同材料进行电极装配所得LSV表征结论(c)[44];*H在不同催化材料中Gibbs自由能计算结果(d)[48]Fig.5 Schematic illustration for the synthesis of CoNiP@NiFe−LDH hierarchical arrays (a);Photographs of water−splitting system(b);LSV results of two−electrode cell assembled by various materials(c)[44];The*H Gibbs free energy of different catalysts(d)[48]
1.4 光电协同催化分解水产活性氢/氧
LDHs 材料在光电催化分解水领域均有较为广泛的应用,考虑到LDHs 独特的二维层状结构[49],因而也常常通过光电沉积等方式与金属氧化物、钙钛矿材料等相结合制备异质纳米结构,通过协同作用提升其催化性能。Duan等[50]将ZnFe−LDH 通过光辅助电沉积方式与TiO2相结合,将ZnFe−LDH 纳米阵列垂直生长并高度均匀分布于TiO2表面[图6(a)],有效提升了TiO2材料的空穴传导效率,通过该方式制备得到的复合催化材料光电OER 催化性能相较于纯TiO2材料有明显提升[图6(b)],实现了光能高效转化为化学能的效果。通过DFT 计算可知,该材料性能显著提升可归因于层板间相互作用的增强和ZnFe−LDH 及TiO2两种不同材料能带结构的高匹配度,使复合材料的光生电子分离效率及光子的注入效率均有显著提升。该材料制备的光阳极在工作过程中,光生空穴可高效地由TiO2迁移至ZnFe−LDH 进而提升该过程中水氧化速率[图6(c)]。该课题组进一步通过将Ag 纳米粒子及ZnFe−LDH 材料作为助催化剂负载于WO3表面实现了对自然水资源(如海水等)直接光电分解制备氢能源,使得LDHs材料光电协同分解水应用有望拓展到工业生产领域[51]。在此基础上,Feng 等[52]通过电化学沉积将还原TiO2材料与Co基LDH材料相结合,通过对复合材料能带结构的进一步修饰,提高光生电子空穴的分离效率,促进水分子分解形成*O中间体进而释放出O2,因而实现了光阳极催化性能的大幅度提升。

图6 TiO2/ZnFe−LDH−PE 复合催化剂合成过程示意图(a);复合催化剂光阳极J-V曲线(b);催化过程中材料水氧化过程示意图(c)[50];ZnO@CoNi−LDH 核壳结构纳米阵列制备示意图(d);电沉积过程总时间对光电转换效率影响(e);ZnO@CoNi−LDH光催化水氧化催化过程机理示意图(f)[53]Fig.6 Schematic illustration for the fabrication of TiO2/ZnFe−LDH−PE NAs(a);J-V curves(b);Schematic illustration for the PEC water oxidation process over the TiO2/ZnFe−LDH photoanode(c)[50];Schematic illustration of the fabrication of ZnO@CoNi−LDH core−shell NWs array(d);IPCE for ZnO@LDH electrode with various LDH deposition time(e);Schematic illustration of the photoelectrochemical water oxidation process by the as−obtained ZnO@CoNi−LDH core−shell NWs array (f)[53]
Duan 等[53]同样通过将半导体材料与LDHs 相结合方式合成高转化效率的光阳极,将CoNi−LDH 通过电化学沉积在ZnO 纳米线阵列上,制备得到具有核壳结构的ZnO@CoNi−LDH [图6(d)],通过控制沉积时间对材料的光电转换效率进行优化[图6(e)],该光阳极在工作过程中显著提升了光生载流子的分离和利用效率,采用电场调整氧化电位,通过ZnO与助催化剂CoNi−LDH 对催化OER 过程的协同作用,实现了高效催化分解水制备氧气[图6(f)]。为了进一步优化反应性能,Ma 等[54]将钙钛矿材料LaFeO3引入该体系中,制备得到CoAl−LDH/LaFeO3/ZnO 纳米光阳极材料,利用ZnO 与LaFeO3之间形成的异质结结构,进一步提高光生载流子的分离效率。
1.5 光催化水氧化合成过氧化氢
作为一种强氧化剂,H2O2被广泛用于化学合成、医疗、食品和环境治理等领域[55]。目前H2O2工业生产方法以蒽醌法为主,该方法成熟且易于大规模生产,但是面临着选择性低、工艺复杂、耗能大等问题[56]。因此,以价格低廉的水和氧气为合成原料是一种绿色且经济的合成方法,可以实现在避免使用有机试剂或贵金属催化剂的前提下,在温和条件下高效合成H2O2。该方法包括水氧化和氧还原两个反应,因此设计出具有双功能的催化剂是高效合成H2O2的关键。很多含有过渡金属的混合金属氧化物(MMO)对于水氧化具有优异的催化效果[57−59],而LDHs经焙烧后得到的MMO兼具有活性组分高分散的优势,有利于活性位点的暴露,因此,通过合理的化学组分调控,可以得到具有双功能的高催化活性的H2O2合 成 催 化 剂。2016 年,Xiang 课 题 组[60]以NiFe−LDH 为前体制备出MMO@C3N4光催化剂。该材料较MMO 具有更多的活性位点,且能带整体正移,更有效地推动了水氧化反应的顺利进行[图7(a)],兼具催化氧还原和水氧化反应的功能,能够在无牺牲剂的条件下光催化合成H2O2,且最高浓度可达63 μmol·L−1[图7(b)]。该课题组[61]随后继续以ZnTi−LDH 为前体煅烧得到ZnTi−MMO 用于光催化水氧化合成H2O2。在反应过程中,ZnTi−MMO 发生拓扑转变,转化为TiO2−ZnTiO3,H2O2生成速率增大[图7(c)],而H2O2分解速率显著减小[图7(d)],催化性能极大提高,H2O2浓度可以达到超过70 μmol·L−1。LDHs 及衍生材料用于光合成H2O2,丰富了H2O2的清洁可持续生产途径。

图7 C3N4和MMO@C3N4的能带结构和电荷转移示意图(a);MMO@C3N4、Ni@C3N4、Fe@C3N4和MMO/C3N4−Mix光合成H2O2的浓度−时间曲线(b)[60];TiO2−ZnTiO3、ZnTi−MMO和P25光合成H2O2的浓度−时间曲线(c);H2O2在TiO2−ZnTiO3、ZnTi−MMO、P25上的分解曲线(d)[61]Fig.7 Scheme of energy levels and charge transfer pathways of C3N4 and MMO@C3N4(a);The light−driven H2O2 generation in O2−equilibrated conditions over MMO@C3N4,Ni@C3N4,Fe@C3N4,and MMO/C3N4−Mix(b)[60];The light−driven H2O2 generation(c);H2O2 decomposition over TiO2−ZnTiO3,ZnTi−MMO and P25(d)[61]
2 LDHs 基纳米材料应用于催化加氢及氧化反应
催化加氢还原/氧化过程在有机反应中应用极为广泛,是生产各种化工原料的一种常用的转换方式,涉及含碳化合物的催化加氢还原及氧化过程需首先经历化学键的活化断裂过程,而因多数含碳化合物中C—O 或C—C 键键能较高,因此反应需要较为苛刻的外界条件(如高温高压)[62−63],制备合适的催化剂以促使该类反应得以在温和条件下进行是研究的难点。LDHs 及其衍生物因其电子结构以及金属元素高分散的特性,在含碳化合物催化加氢及氧化过程中具有广泛应用。
2.1 LDHs 基催化剂应用于光驱动的CO 加氢制备短链烯烃过程
短链烯烃在聚合物合成及药物合成等方面都有极为重要的应用,而短链烯烃的常规合成方法需要苛刻的反应条件,同时消耗大量的化石能源[64]。采用合成气(CO 与H2)制备烯烃是目前最常用的合成方式[费托反应(FTO)过程],在该反应过程中,Ni、Co、Fe 基催化剂通常可催化C O 键断裂的同时促进C—C 键形成,在催化过程中具有高转化率和选择性,被认为是该过程的一类理想的催化剂[65−66]。LDHs 因其层板金属元素种类可调的特性,Ni、Co、Fe 基LDHs 材料经焙烧还原处理可作为催化FTO 过程的一类理想的催化材料。传统制备合成气的方法通常采用热催化方式,如Jong等采用Co基催化剂材料[67]及Fe 纳米粒子材料[68]通过热催化制备合成气,Ma等[65]采用Fe基催化剂材料在热催化条件下实现合成气的高选择性催化转化。近期,Wei 等[69]以CuMgFe−LDH 为前体,通过焙烧结构拓扑转变过程及活化过程获得Fe5C2纳米团簇负载的Cu 纳米粒子催化剂材料,该材料可实现低压1 MPa 条件下催化CO转化为长链醇类物质,大大降低了固有的热催化过程对反应条件的苛刻要求[图8(a)]。通过原位CO−TPD 表征证实,该材料中焙烧产物Fe5C2及Cu的界面结构对该过程性能有显著影响[图8(b)]。对于合成气转化过程的反应探究从未止步,Zhang等[70]以含有Ni元素的LDHs 材料焙烧还原过程得到Ni/NiO基材料,利用金属Ni 与NiO 材料形成的界面微系统,有效调节了催化剂材料中Ni 原子的电子结构,实现了对FTO 过程光驱动高选择性催化。该界面微系统相比于纯相NiO 或Ni 结构,高碳烃的选择性由8%提升至60%。该课题组通过电子自旋共振(ESR)测试进一步研究了反应的机理[图8(c)]:在光驱动反应条件下催化剂产生载流子,进一步活化了CO 以及H2分子,该机理为研究CO 与H2太阳能驱动催化转化提供了思路,为后续水分解耦合氧化/加工还原制备高附加值化学品提供了理论支撑。为了进一步拓展该材料在催化领域的应用,该课题组[71]将该过程的催化剂材料的选择拓展到光驱动的Co基、Fe 基LDH 材料,通过DFT 计算探讨该系列催化剂导致FTO 过程性能大幅提升与催化剂的结构密不可分[图8(d)]。该课题组通过对ZnCoAl−LDH [图8(e)]在不同温度下焙烧还原,得到一系列Co 基催化剂材料,在焙烧还原过程中生成的ZnO 和Al2O3在提升该催化剂材料结构和热力学稳定性的同时,也提高了Co 材料的催化还原效果。通过多种氧化物的有机高效协同作用,产物中短链烯烃选择性可达36%。EXAFS 表征结论表明不同焙烧温度下Co 的主要存在形式为Co 和Co3O4,证实最佳催化效果条件下Co 元素的存在状态主要为Co/Co3O4。Fe 元素因为其优越的电子结构,被认为是在CO 催化加氢过程中更为有效/廉价的一种金属元素,该课题组将ZnFeAl−LDH 在不同温度下还原焙烧产物同样应用于CO 催化加氢过程,通过对催化剂电子结构的可控调变,500℃下还原焙烧产物——Fe−FeZnOx催化剂在光驱动的CO 加氢过程中短链烯烃选择性可高达43%[72]。此外,通过以LDHs 材料为前体制备的CoFe 合金,同样可以实现光热催化CO2高选择性生成高碳烃的催化效果[73]。
2.2 LDHs基催化剂用于光/电驱动醇氧化过程

图8 四种不同Cu/Fe比例下生成醇类物及长链醇类物转化率(a);Fe1、Cu4及CuxFey CO−TPD表征结果(b)[69];Ni−NiO结构的电子自旋共振−结果(c)[70];DFT计算Fe3O4、4O/Fe和4O/Fe3Zn光催化剂催化过程CO2生成,C2H4吸附及氢化能量势垒(d);ZnCoAl−LDH 为前体H2 300~700℃还原制备Co基催化剂过程示意图(e)[71]Fig.8 Alcohols STY at different pressures over catalysts with four Cu/Fe ratios (1/1,2/1,4/1,and 6/1)(a);CO−TPD profiles of Fe1,Cu4,and CuxFey samples(b)[69];ESR spectrum over Ni−NiO structure(c)[70];The potential energy profiles for CO2 formation,C2H4 adsorption,and hydrogenation under excited states on Fe3O4,4O/Fe,and 4O/Fe3Zn(d);Fabrication of Co−x catalysts by H2 reduction of ZnCoAl−LDH nanosheets at 300—700℃(e)[71]
醇类化合物的氧化过程也是化工生产过程中一类重要的有机反应。乙醇因其具备高能量密度、低毒性、易于储存等多重优势引起广泛重视。LDHs基催化剂因其层状结构的特性、材料的比表面积较大、在多种醇类物质中稳定性良好且具有较好的电催化性能,因此作为醇类氧化过程的催化剂材料得到了广泛的应用[74−75]。制备具有合适的孔道结构及高比表面积的LDHs 基材料,是提升其醇类物质氧化性能的重要途径。Duan 等[76]通过在合成过程中添加表面活性剂,可控制备了MgFe−LDH 微纳米球结构[图9(a)],在很大程度上提高了催化剂的比表面积,实现了孔径分布的调控[图9(b)],提高了催化过程中的电活性材料对于法拉第还原过程的效率,同时加快了催化过程中的传质效率,相对于卵壳结构或实心结构催化剂,该催化剂应用于碱性燃料电池的乙醇催化过程时,展现出了更优越的催化性能[图9(c)]。该催化剂材料的设计也为制备其他类型微纳米球结构的催化材料提供了发展思路和可能路径。Zhang 课题组[77]选用Au 纳米团簇负载的LDHs 基催化剂材料,实现了醇类物质氧化性能的高效提升。该课题组采用水溶性谷胱甘肽修饰的金纳米团簇为前体,与Al基LDHs材料复合,通过N2气氛下焙烧过程,得到粒径小于1.5 nm 的金团簇−LDHs 基复合材料,其中金纳米粒子在LDHs 基底表面高度均匀分散[图9(d)]。在甲苯溶剂中该材料将苯乙醇转化为乙酰苯的转化效率可达99%,其转换效率(TOF)可达46500 h−1,且可拓展底物至多种醇类物质,极大促进了对醇类物质催化氧化过程的发展和探索。类似地,将直径小于5 nm 的Au 纳米粒子负载于LDHs表面[78],实现了对醇类材料在无外加氧化剂下,脱氢制备羰基化合物,并在该过程中实现了接近百分之百的产物选择性[图9(e)]。
3 光电分解水及催化加氢/氧化反应耦合

图9 MgFe−LDH可控制备花样形貌微球结构催化剂过程示意图(a);N2吸脱附测定MgFe−LDH纳米微球结构比表面积与孔径分布(内置图)(b);MgFe−LDH微纳米球结构修饰电极伏安特性曲线(c)[76];Au负载Al基LDH焙烧制备过程示意图(d)[77];Au/HT催化剂通过DP法催化醇类化合物脱氢生成羰基化合物效率(e)[78]Fig.9 Schematic illustration of the morphological evolution process of the as−obtained flower−like hierarchical LDH microspheres(a);N2−sorption isotherms and pore size distribution(inset)of MgFe−LDH microspheres with different inner architecture(b);Cyclic voltammograms at the MgFe−LDH microspheres modified electrodes(c)[76];Design schematic of the Au−NCs/LDH catalyst(d)[77];Time course for the dehydrogenation of benzyl alcohol over Au/HT catalyst prepared by the DP method (e)[78]

图10 水分解及有机催化反应耦合反应设计思路Fig.10 Water splitting combined with hydrogenation/oxidation for the synthesize of high−valuable fine chemicals
光电分解水过程时会产生大量*H 及*O 中间体,而工业常用催化加氢及催化氧化过程涉及*H及*O的消耗,为简化反应过程同时提高原料及能量的利用效率,将*H 及*O 中间物种作为反应的有效中间纽带,通过调控反应条件和优化催化剂结构,实现两种反应的有机耦合,将水分解产生的活性物质直接应用于催化加氢还原/氧化过程,提高反应催化转化效率的同时可减少能量的损耗。目前LDHs 基材料、修饰后的金属有机框架(MOF)材料[79−80]、CdS/石墨烯材料[81]、C3N4材料[82]等多类催化剂在光电分解水及催化加氢/氧化反应等领域都取得了显著的成效。基于LDHs 在工业催化加氢还原及氧化过程和光电分解水等多个领域的发展,对于以上两种反应的耦合,如水分解及工业加氢还原、氧化过程有机耦合,制备高碳烃、苯酚等多种精细化工产品具备优异的催化性能(图10),将在以下进行详细的讨论。
3.1 水分解与催化加氢还原反应耦合
水分解及加氢耦合目前已经广泛应用于CO2还原过程中[83−84]。Zhang等[85]通过反向微乳液法可控制备尺寸小于30 nm 的超薄ZnAl−LDH,将该ZnAl−LDH 应用于CO2还原制备CO 过程,相比于块体ZnAl−LDH 在选择性和转化率上都显著提升。通过DFT 计算可知,该超薄ZnAl−LDH 对于CO2及H2O 吸附强于块体材料。由X射线吸收精细结构谱(XAFS)表征结果可知,材料中存在O空位及晶格畸变,提供了大量不饱和位点。电子自旋共振结果表明,超薄ZnAl−LDH 中存在Zn+−VO。反应过程中水蒸气在催化剂的作用下裂解产生的*H 直接用于CO2还原过程,可以明显提升光催化CO2还原的性能。近期,本课题组[86]通过外加甲酰胺的方式,一步制备合成了单层NiAl−LDH,该合成产物尺寸约30 nm,厚度小于1 nm。单层NiAl−LDH 在水参与反应下,在波长大于600 nm 时,其光催化CO2还原反应中可完全抑制副产物H2的生成[图11(a)],并且高选择性地合成了含碳产物(CH4和CO)。单层NiAl−LDH 材料中丰富的金属缺陷和氧缺陷等不饱和位点[图11(b)],可作为催化活性位点,因而实现了性能的大幅提升。进一步通过DFT 理论计算发现其完全抑制氢气生成原因是:在光照大于600 nm 的范围内,光敏化剂中电子只可以获得能量被激发到第一单线激发态(−1.008 eV)或第一三线激发态(−0.955 eV),低于产氢最低价带能级,不足以驱动反应过程中产生的*H还原为H2分子[图11(c)]。同时,DFT 计算结果也证实单层NiAl−LDH 相较于块体NiAl−LDH 显著降低了CO2催化还原为CH4的Gibbs 自由能,使得在大于600 nm光照下光敏化剂中电子可以满足生成CH4的能量差。材料的缺陷及元素空位可显著提升材料催化性能,Zhang 课题组[87]通过在Co3S4−x材料中引入S 空位,实现了芳香硝基化合物在低电位下高效转换为芳香氨基化合物的效果。在该催化过程中,通过氘(D)元素取代证实,催化加氢过程中H 元素直接来源于材料中外加的H2O,而S 空位在反应过程中的主要作用为促进*H 中间体的高效形成,证实了*H 在该催化过程中的重要作用[图11(d)]。类似地,该课题组[88]用D 代方式对Pd−P 作为阴极材料催化炔烃的部分氢化过程亦进行了反应过程中H 元素的来源及过程机理研究,在该催化剂的作用条件下,实现了对炔烃部分氢化过程高达99%的选择性[图11(e)]。

图11 单层NiAl−LDH在不同波段光照CO2还原产CH4、CO及H2选择性对比(a);缺陷单层NiAl−LDH层板结构示意图,VM代表材料中金属缺陷(M=Ni,Al),VOH 代表材料中OH缺陷(b);光敏化剂Ru(bpy)3Cl2 单线态及三线激发态能级及单层NiAl−LDH中CBM、缺陷态、VBM相对于NHE能级关系(c)[86];以CoS纳米片作为光阴极S空位还原氢化芳香硝基化合物生成功能性芳香氨基化合物过程示意图(d)[87];以Pd−P为阴极,以H2O(D2O)作为H源或D源对炔烃进行部分氢化过程示意图(e)[88]Fig.11 Selectivity of CH4,CO,and H2 in CO2PR on monolayer NiAl−LDH under different wavelength(a);Illustration for the species of defects on monolayer NiAl−LDH.VM represents metal defect(M=Ni,Al),VOH represents the hydroxyl defect(b);The energy levels for the singlet and triplet excited states of photosensitizer Ru(bpy)3Cl2,together with the band edge placements for the CBM,defect state,and valence band minimum(VBM)of monolayer NiAl−LDH(VNi&OH)versus the normal hydrogen electrode(NHE)(c)[86];Illustration of sulfur vacancy−promoted selective synthesis of functionalized aminoarenes via transfer hydrogenation of nitroarenes with H2O as the hydrogen source over a cobalt sulfide nanosheet cathode(d)[87];Illustration of selective transfer semihydrogenation of alkynes with H2O(D2O)as the H(D)source over a Pd−P cathode(e)[88]
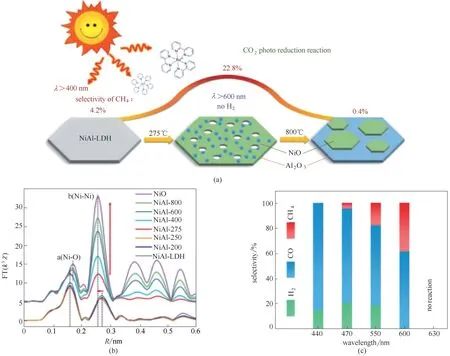
图12 NiAl−LDH经焙烧拓扑转变形成高缺陷浓度NiO结构催化CO2还原示意图(a);NiO,NiAl−x,NiAl−LDH 的EXAFS R空间图(b);NiAl−275在不同波长的单色光下催化CO2还原选择性对比(c)[89]Fig.12 Schematic illustration of defect−containing NiO derived from the topological transformation of NiAl−LDH(a);The corresponding k3−weighted FT spectra of NiO,NiAl−x,and NiAl−LDH(b);Selectivity of NiAl−275 under different monochromatic light(c)[89]


图13 CoFe−LDH及NiCoFe−LDH 材料室温下光致发光光谱图(a);波长>500 nm条件下CoFe−LDH与NiCoFe−LDH 催化CO2还原性能对比(b)[90];不同种类Al基LDHs紫外可见光谱(c);不同种类Al基LDHs可见光催化CO2生成CO,H2及CH4还原性能对比(d)[91]Fig.13 Room−temperature photoluminescence(PL)spectra of CoFe−LDH and NiCoFe−LDH(a);Selectivity of CH4,CO,and H2 under irradiation above 500 nm for CoFe−LDH and NiCoFe−LDH(b)[90];UV−vis spectra for the various u−MAl−LDH photocatalysts(c);Production rates of CO,H2,and CH4 on various u−MAl−LDH photocatalysts in CO2PR under visible light(d)[91]
此外,不同比例的合成气(CO 和H2)是重要的工业生产原料,常被用于制备烃类及多种高附加值的精细化学品等[92]。LDHs 材料用于CO2还原制备合成气,可实现其大范围比例的调控[93],本课题组[94]利用CoAl−LDH 及MoS2材料复合形成的异质结结构,通过调变反应物中所加催化剂的浓度[图14(a)],即可实现H2和CO 生成比例在1.3∶1~15∶1 的调变[图14(b)],该工作采用较为简单的方式即可实现CO2的高效转化。本课题组[95]研究发现,对于LDHs 材料,插层阴离子的种类可影响层板的电子结构和缺陷,进而影响CO2还原反应的选择性,通过对比CO及N插层的NiAl−LDH 在CO2还原过程中对于产物CH4选择性[图14(c)],发现N插层的NiAl−LDH 因存在更高程度的O 缺陷,具有更高的CH4选择性。稀土金属的负载亦被证实可以显著调控催化材料的电子结构进而影响其催化性能,不同浓度的CeO2负载LDH纳米结构被证实对于CO2还原过程产物选择性有明显影响[96],在由低到高不同CeO2浓度负载的情况下,MgAl−LDH 对于CO2还原过程CO/H2还原产物比例整体呈火山型规则,其中Ce 负载量为0.15%时,其产物CO/H2比值约为1/1.3,产物CO 选择性达到最高[图14(d)]。
水分解不仅可与CO2还原过程耦合,同样可应用于耦合芳香胺功能化取代[87]或者固氮过程等。Zhang 等[21]合成含有大量O 缺陷的CuCr−LDH,丰富氧缺陷的存在导致LDH 材料中具有明显的正六面体结构畸变,在水存在条件下,CuCr−LDH 可高效地光催化将N2转化为NH3,使得催化性能相比于普通块体CuCr−LDH 有明显提升,相较于其他种类LDHs材料也有着明显的性能优势。该催化体系通过催化水分解产*H 过程与N2还原过程的耦合,在25℃及λ>500 nm 光照的温和反应条件下,N2到NH3的转化效率可达7.1 μmol·L−1。原位红外表征结果证明在整个催化过程中,反应体系的N2分子在催化剂的表面与H2O 分子催化转化形成NH+4,实现了CuCr−LDH 表面的高效固氮,为高效光催化分解水耦合N2还原固氮提供了思路。

图14 不同浓度LDH/MoS2催化剂调控合成气比例示意图(a);不同LDH/MoS2催化剂浓度催化CO2还原反应产率及选择性(b)[94];不同插层阴离子NiAl−LDH对于CO2还原过程产物选择性示意图(c)[95];CeO2不同比例负载MgAl−LDH催化选择性(d)[96]Fig.14 Schematic illustration of photocatalytic CO2 reduction to tunable syngas on CoAl−LDH/MoS2 heterostructures with different catalyst concentrations(a);LDH/MoS2 nanocomposite yield and selectivity of CO and H2 in CO2PR with different concentrations(b)[94];Schematic diagram of the selectivity of photocatalytic CO2 reduction by different anion intercalated NiAl−LDH(c)[95];Selectivity of LDH and Ce−x(d)[96]
3.2 水分解与催化氧化过程耦合
水分解过程产生*H 的同时伴随有大量*O 的生成,通过水分解过程产生的*O中间体与催化氧化过程耦合,可达到温和条件下进行催化氧化过程的目的。基于可再生能源高效利用与绿色化工的重大需求,针对光电解水耦合绿色化工加氢/氧化过程,北京化工大学化工资源有效利用国家重点实验室Duan 等[97]首次报道了一种光电解水耦合氧化反应路径,通过复合TiO2纳米纤维、石墨碳层材料及Co3O4制备光阳极材料,耦合了水分解过程及苯甲醇氧化过程,实现了室温下催化苯甲醇氧化至苯甲醛,在TiO2、TiO2/C、TiO2/C/Co3O4三种催化材料中,TiO2/C 展示出了最优越的转化率及选择性,其选择性可达99%。在温和催化条件下反应速率可达76 mmol·g−1·h−1,整体反应速率远超过文献中已有的单独的光催化过程的反应速率[图15(a)]。为探究过程机理,该课题组通过DMPO自旋电子猝灭过程证实,TiO2/C/Co3O4材料中存在更丰富的·O2−[图15(b)],是性能提升的关键影响因素之一。根据电子自旋猝灭表征推断,该过程机理主要分为两个步骤[图15(c)]:光照条件下电子空穴对在TiO2中生成,电子进一步转移至石墨材料的同时,空穴则迁移至Co3O4材料,该电子及空穴的高效分离促进了性能的大幅提升。该课题组[98]进一步提出将水分解过程与尿素的氧化过程有机耦合,通过电化学沉积与气相沉积等方法,合成了SnO2@BiVO4/Co−Pi 复合催化剂,该耦合过程催化性能在催化尿素氧化过程中性能优于BiVO4材料[图15(d)]。
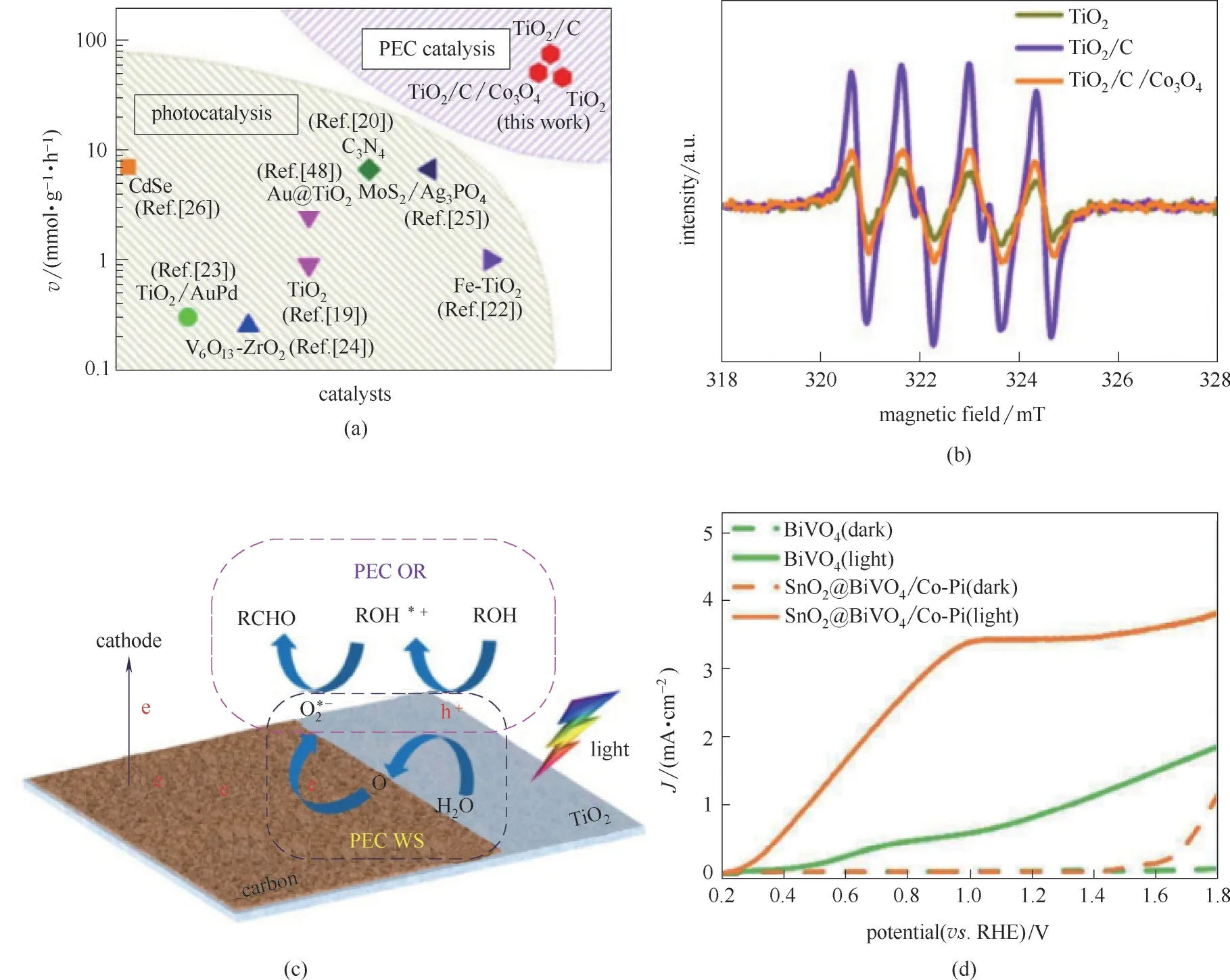
图15 本文中PEC催化速率与前人光催化过程反应速率对比(图中文献号是参考文献[97]中的文献号)(a);DMPO自旋电子捕获剂检测TiO2、TiO2/C和TiO2/C/Co3O4中·O2−结果(b);PEC WS−OR耦合氧化过程示意图(c)[97];SnO2@BiVO4/Co−Pi光照条件下催化尿素氧化光电流曲线(d)[98]Fig.15 Comparison of reaction rate between PEC catalysis in this work and photocatalysis reported previously (the Ref.numbers in the figure were Ref.numbers of the Ref.[97])(a);DMPO spin−trapping ESR spectra recorded for DMPO−·O2−over TiO2,TiO2/C,and TiO2/C/Co3O4 sample,respectively(b);Schematic illustration for the PEC WS−OR coupling process(c)[97];Photocurrent−potential curves under illumination of urea oxidation(d)[98]
不仅是复合催化材料,LDHs 材料在耦合水分解过程也展现出了优越的催化性能,本课题组[99]合成了具有丰富氧缺陷的ZnTi−LDH 结构[图16(a)],将水分解过程与苯氧化过程进行有机耦合,实现苯到苯酚选择性氧化高达87%的转化效率。该LDHs 材料能带结构与苯催化制备苯酚过程高度相符[图16(b)],使得ZnTi−LDH 材料在耦合水分解与苯氧化过程中,具有高效的性能[图16(c)]。根据不同气氛该催化剂对苯催化氧化过程的探究证实,在氮气氛围下,该氧化过程反应效率大幅降低[图16(d)],证实水分解产生活性氧对苯选择性氧化过程有明显促进作用。为进一步探究该催化过程中间产生的自由基中间体,采用DMPO作为自旋捕获剂,通过ESR测试证实反应中存在·OH、·O2−[图16(e)、(f)],说明ZnTi−LDH 催化苯氧化过程中活性氧中间体至关重要。该系列工作首次利用光电解水耦合催化氧化,在精细化学品氧化以及尿素氧化方面展现了较单纯光、电催化等催化方式更优异的催化性能,为光电解水耦合氧化/加氢提供了催化剂设计的思路。
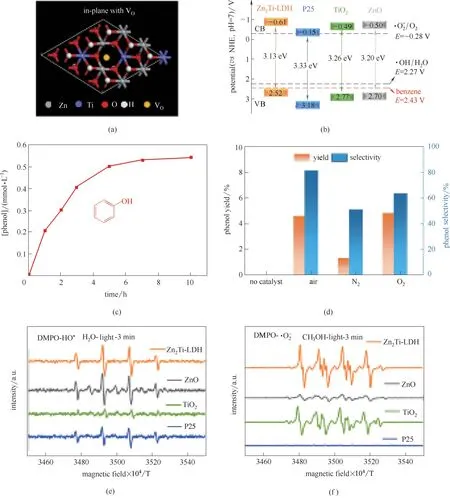
图16 存在O缺陷的ZnTi−LDH材料结构示意图(a);ZnTi−LDH能带结构示意图(b);苯酚产率结果(c);催化剂及反应气氛调控对苯酚产率影响(d);DMPO自旋捕获以检测·OH(e);DMPO自旋捕获以检测·O2−(f)[99]Fig.16 Supercell model of ZnTi−LDH layer doped with VO vacancies(a);Schematic band diagrams(b);Reaction time profiles of phenol(c);Conversion of benzene oxidation(d);5,5−dimethyl−1−pyrroline N−oxide(DMPO)as spin−trapping agent under UV−vis light irradiation to detect·OH(e)and·O2−(f),respectively[99]
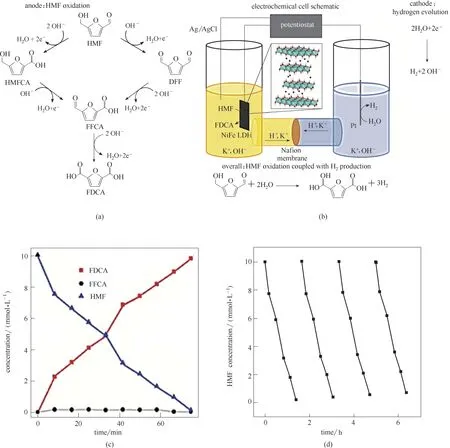
图17 HMF氧化流程及中间产物(a);糠醛氧化耦合水分解催化过程电解池示意图(b);催化过程中HMF及其氧化产物浓度随时间的变化(c);NiFe−LDH生长于碳纸表面作为催化材料的LSV测试结果(d)[102]Fig.17 HMF oxidation process and products(a);Schematic diagram of the electrochemical system used for the overall cell reactions(b);Concentration changes of HMF and its oxidation products with the time of chronoamperometric tests (c);LSV curves of the NiFe−LDH nanosheet growth on carbon fiber paper(d)[102]
通过改良电解池耦合糠醛(HMF)氧化过程可有效提高电催化水分解效率[100],同时有效避免在水分解过程中产生的*O对电解池隔膜结构的影响[101],延缓电解池老化,有效延长其使用寿命。Huber 等[102]采用水热法将NiFe−LDH 原位生长于碳纸材料表面,作为电催化HMF氧化过程的阳极材料。通过设计新型电解池将HMF 选择性氧化过程与水分解过程耦合,该水分解过程产生的活性氧中间体可直接用于糠醛氧化过程[图17(a)、(b)],在碱性水相体系中,对氧化产物呋喃二甲酸的产率为98%,选择性高达99.4%[图17(c)]。根据LSV测试结果表明,水分子的存在可有效促进糠醛的氧化过程,同时糠醛的存在对水分解过程也有反向促进作用,将糠醛加入水分解体系中可使该耦合过程过电势显著降低[图17(d)],实现了水分解过程与糠醛氧化过程高效有机耦合。
4 结论与展望
氢能的高效率低成本制备是目前的研究热点,目前多数H2制备及进一步利用过程通常为独立的两个反应过程,本文综述了通过利用光电分解水耦合精细化学品加氢还原/催化氧化制备高附加值精细化学品的进展,为温和条件下化学品的制备提供思路。但决定该耦合过程效率的关键在于选择合适结构的催化剂材料。
LDHs 因层板组成金属具有丰富的可调变性、层间客体可调、形貌尺寸可控等优点,研究人员可以进一步调控LDHs 基催化材料的能带结构、电子结构、缺陷结构,进而实现对多种反应的高效催化转化。且通过调控LDHs 的形貌尺寸等因素,可大大提升对某类特定反应的选择性。此外,LDHs 材料具有原料丰富、易于规模化合成等诸多优点,近些年其在光电等催化领域得到了长足发展,在诸如水分解、CO2还原、有机物氧化等多种催化反应中都体现了优越的性能。利用LDHs 本身层板组成元素高度分散的特性,以LDHs 材料为前体,经过拓扑转变合成的氧化物、氮化物、磷化物等及多种催化材料,在光电热等多领域取得了广泛的应用。本文着眼于LDHs 基材料在光电领域的重要应用,并通过光电分解水产*H 与*O 这一重要的中间活性物质耦合不同反应,实现催化还原/催化氧化过程与水分解过程的高效有机耦合。该耦合过程不仅可以避免多步反应导致的能量及反应原料损耗,而且可以实现反应过程中催化转化效率的显著提高,体现了绿色化学的发展理念。
不可否认的是,LDHs 在催化领域的多方发展以及水分解与氢化/氧化反应耦合过程中依然存在很多尚待解决的问题:(1)反应的普适性。LDHs基催化剂用于水分解及氧化/加氢过程的耦合仍待进一步拓展至更多类型的反应,目前的反应主要局限于CO2还原制备合成气/烃类,固氮过程及醇类醛类物氧化过程。(2)副产物高效分离。部分催化过程选择性仍难以接近99%,存在副产物难以分离等问题。(3)反应效率仍需要提高。相比于传统热催化,光电催化的效率仍相对较低,距离实际工业应用仍然有一段距离。(4)机理研究还需更加明确。虽然通过电子顺磁共振、原位红外、瞬态光谱等手段,可以捕获反应部分中间体,但反应机理尚不完全明确,仍缺乏有效的原位表征技术对机理进行瞬态/实时的研究。相信在不久的将来,随着研究的进一步深入,高端仪器的开发设计以及市场化,上述问题能得以解决,实现能源可持续发展的终极目的。
