不同背膘厚猪肠道微生物多样性差异的比较
2020-09-25王秀娜王春旭高安崇白春艳孙博兴
王秀娜,王春旭,高安崇,梁 爽,白春艳,孙博兴*
(1.吉林大学 农业实验基地,吉林 长春130061;2.吉林大学 动物科学学院,吉林 长春 130062)
猪的肠道微生物在调节宿主脂肪代谢等生理机能方面发挥着重要的作用[1]。研究表明,猪的平均日增重、饲料转化率和采食量与肠道微生物组成有关[2-5]。据报道,把肥胖的人或小鼠的粪便微生物群移植到无菌或抗生素处理的小鼠体内,导致体内脂肪沉积增加,认为肠道微生物群在调节脂肪代谢中发挥着重要作用[6]。将2种脂肪表型不同的成年金华猪和长白猪的粪便微生物群移植到抗生素处理小鼠体内,发现受体小鼠在脂肪生成特征上与各自的猪供体相似;与金华猪供体相似,受体小鼠肝脏脂质和甘油三酯水平以及脂蛋白脂肪酶活性升高;长白猪粪便微生物群受体小鼠与金华猪粪便微生物群受体小鼠相比关键脂质基因表达增强,血管生成素样-4(angpt-4) mRNA表达减少,这证实肠道微生物群在猪的脂肪代谢调控中起着重要作用[7]。肠道微生物作为调节脂肪沉积的环境因素,对其结构和功能的分析可能成为一种调控猪肉品质新的研究策略。本试验采用高通量测序技术对不同背膘厚猪的肠道菌群微生物的多样性进行比较分析,旨在为通过利用肠道微生物群来调控猪肉品质奠定一定理论基础。
1 材料与方法
1.1 试验动物及样本挑选80头日龄相近、健康状况良好、体质量30~35 kg的军牧1号白猪公猪,测定起始体质量,组建测定群体。所有猪在相同饲养条件下饲养,自由采食和饮水,消毒、免疫等按照猪场饲养管理程序进行。在95~100 kg阶段测定结束体质量,并利用A超测定P2点背膘厚。将测定个体的背膘厚校正到100 kg体质量,校正背膘厚=实测背膘厚×12.83/{12.823+[0.11×(测膘体质量-100)]},日增重=(结束体质量-起始体质量)/饲养天数。根据测定结果,在测定群体中挑选健康、日增重(ADG)无显著性差异,100 kg体质量背膘厚(BF)差异极显著(P=0.000)的高背膘组[ADG=(777.92±54.3) g,BF=(17.6±1.38) mm]和低背膘组[ADG =(758.48±78.85) g,BF=(11.56±1.01) mm]公猪各5头,在同一天收集10头猪直肠处的新鲜粪便,置于液氮罐中保存。
1.2 试验方法取0.25 g粪便样品,用MO BIO PowerFecal®DNA Isolation Kit(MO BIO Laboratories,Carlsbad,CA,USA) 提取微生物DNA,用紫外分光光度计(Thermo Fisher Scientific,USA)进行定量分析和质量判断。选取细菌16S rRNA基因高度可变的V4区用来进行PCR扩增,根据V4区的保守区域进行通用引物设计。forward (P1)5′-AYTGGGYDTAAAGNG-3′;reverse(P2) 5′-TACNVGGGTATCTAATCC-3′。扩增体系(25 μL):5×reaction buffer 5 μL,5 × GC buffer 5 μL,100 mmol/L dNTP 5 μL,10 μmol/L P1 1 μL,10 μmol/L P2 1 μL,DNA Template 2 μL,ddH2O 6.0 μL。PCR反应条件:98℃变性30 s;然后98℃15 s,50℃30 s,72℃30 s,27个循环;72℃再延伸5 min;最后4℃ 5 min结束。利用Axy Prep DNA Gel Extraction Kit对PCR产物进行回收纯化。利用Quant-iT PicoGreen dsDNA Assay Kit对PCR产物在Microplate reader(BioTek,FLx800)上进行定量,然后将每个样品进行等量混合,形成均一的混合物。采用标准的Illumina Tru Seq DNA文库制备实验流程(Illumina Tru Seq DNA Sample Preparation Guide)构建所需的宏基因组上机文库。取1 μL 文库,在Agilent Bioanalyzer机器上用Agilent High Sensitivity DNA Kit对文库做2 100质检。利用Quant-iT PicoGreen dsDNA Assay Kit在Promega QuantiFluor上对文库进行定量,合格的文库计算后浓度应在2 nmol/L以上。对合格的文库,在MiSeq机器上利用MiSeq Reagent Kit V3 (600 cycles)进行2×300 bp的双端测序。
1.3 数据分析利用QIIME软件处理和分析测序序列,调用UCLUST序列比对工具,进行归并和OTU划分。在QIIME软件中使用默认参数,获取每个OTU所对应的分类学信息,同时计算群落丰富度Chao1指数和Shannon指数。使用R软件对Unweighted和Weighted的UniFrac距离矩阵分别进行NMDS分析。使用PICRUSt软件,根据KEGG数据库中微生物代谢功能的类别对群落样本进行预测。
2 结果
2.1 OTU划分和分类地位鉴定运用QIIME软件识别疑问序列,检查并剔除嵌合体序列,最终获得385 719个有效序列。对获得的有效序列按97%的序列相似度进行归并和OTU划分,共得到110 788个OTU,其中HBF组55 550个、LBF组55 238个。各样本的分类地位鉴定结果如表1所示。根据获得的OTU丰度矩阵,使用R软件计算得到HBF组和LBF组共有2 326个OTU,独有的OTU为HBF组1 017个、LBF组783个。

表1 OTU划分和分类地位鉴定结果 个
2.2 Alpha多样性结果如表2所示,LBF组Chao1和Shannon指数均高于HBF组,但差异均不显著(P>0.05),表明LBF组和HBF组微生物群落的丰富度和多样性均无显著性差异。

表2 Alpha多样性分析结果
2.3 Beta多样性Beta多样性分析结果(图1)显示,LBF组和HBF组间微生物群落结构共有33.00%的差异,第1主成分PC1对物种分布的解释量为18.12%,第2主成分PC2对物种分布的解释量为14.88%。
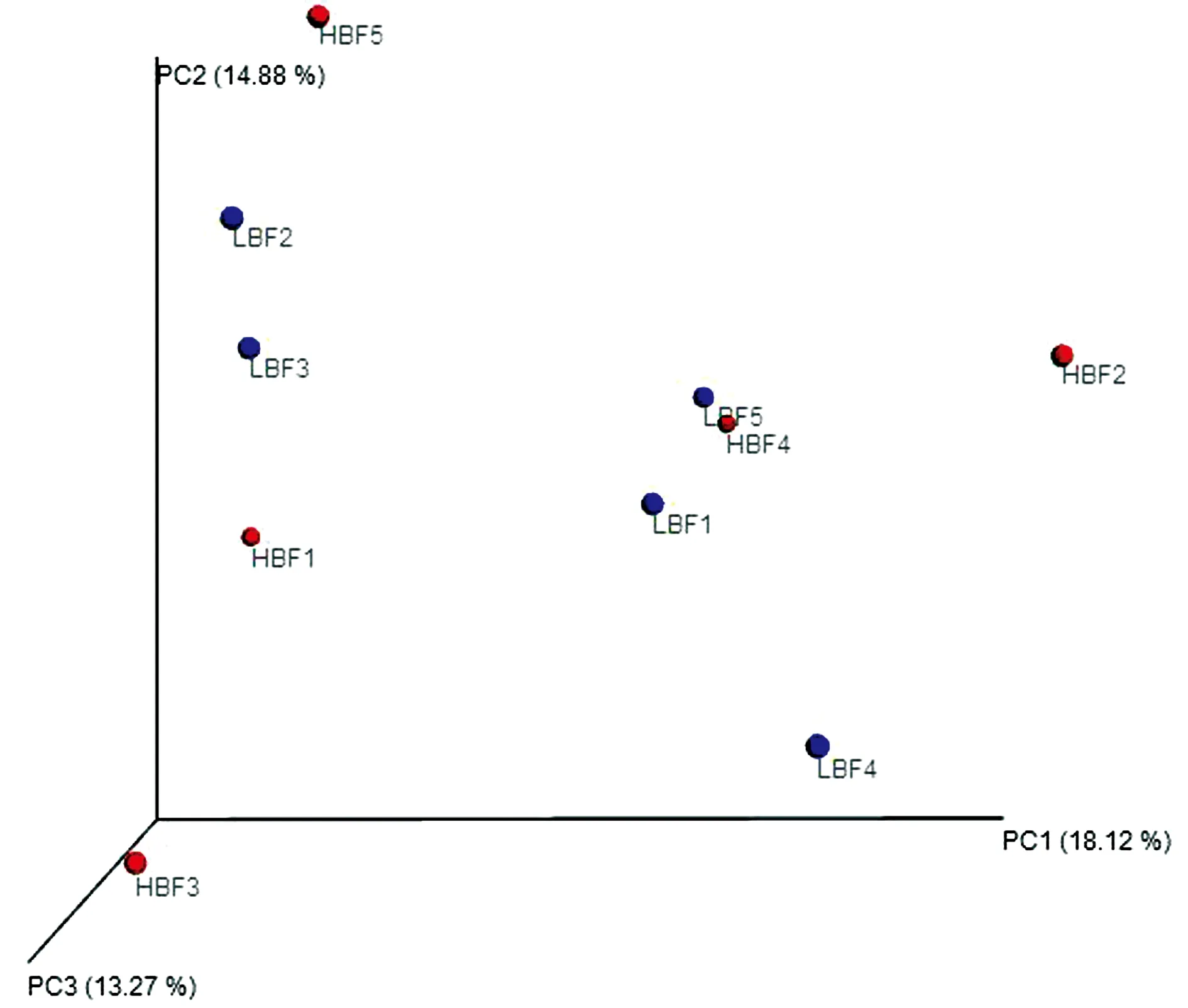
图1 Unweighted UniFrac PCoA分析的样本三维排序
2.4 各分类水平的分类学组成使用QIIME软件,获取各样本各分类水平上的组成和丰度分布。在门水平上,LBF组和HBF组样本中最丰富的门是厚壁菌门、拟杆菌门、变型菌门和螺旋体门,这4个门在所有样品中的丰度均在90%以上。HBF组厚壁菌门的相对丰度高于LBF组7.55%,而HBF组拟杆菌门的相对丰度低于LBF组7.33%,LEfSe线性判别分析表明差异均不显著(图2)。在纲水平上,最丰富的纲是厚壁菌门的梭菌纲和杆菌纲以及拟杆菌门的拟杆菌纲,在所有样本中这3个纲的丰度均为70%~85%。HBF组梭菌纲的相对丰度高于LBF组6.76%,LBF组拟杆菌纲的相对丰度高于HBF组7.30%,LEfSe线性判别分析表明差异不显著(图3)。

图2 各样本在门水平相对丰度分布图
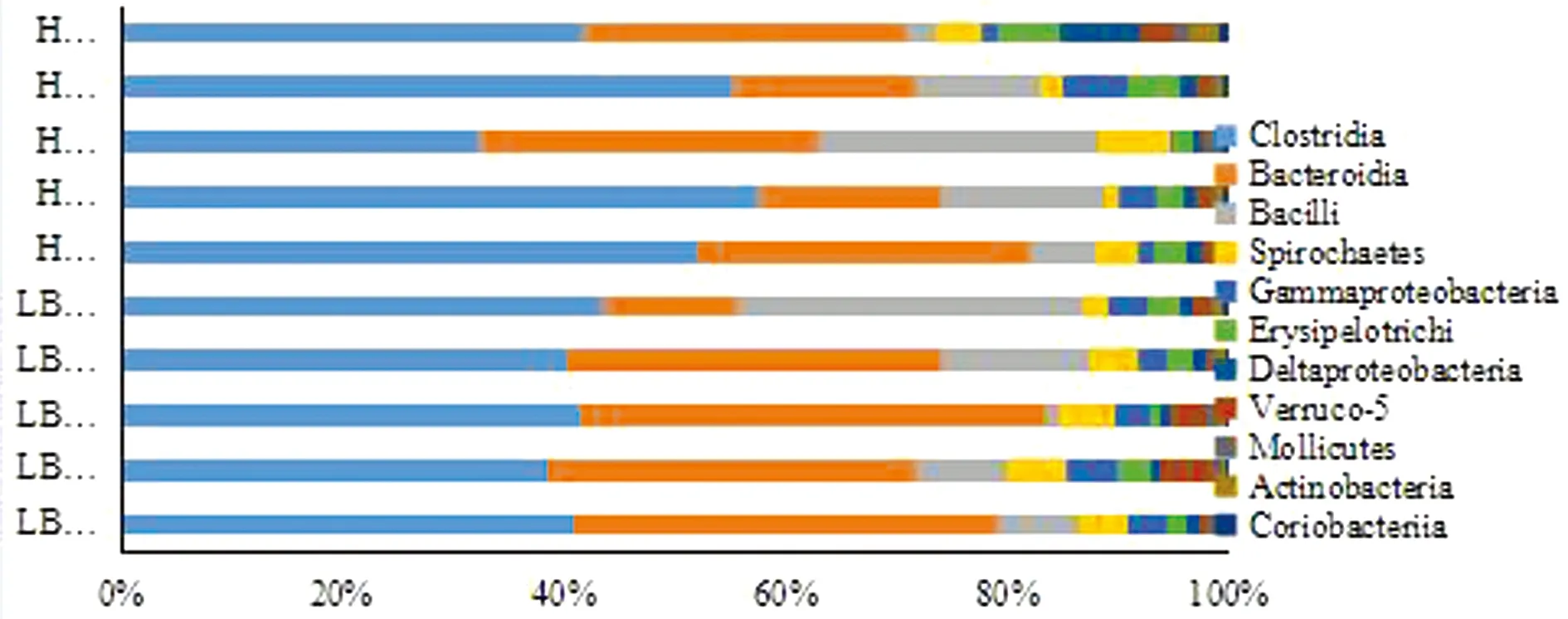
图3 各样本在纲水平相对丰度分布图
2.5 组间分类学组成的差异使用Mothur软件调用Metastats的统计学算法,对门和属水平的各个分类单元在组间的序列量(即绝对丰度)差异进行两两比较,检验结果如图4所显示。在门水平上,HBF组纤维素杆菌门的序列量为1.32%显著(P=0.02,Q=0.08)高于LBF组0.14%;在属水平上,HBF组消化球菌属(P=0.028,Q=0.617)、萨特菌属(P=0.030,Q=0.617)、考拉杆菌属(P=0.035,Q=0.617)、多尔菌属(P=0.042,Q=0.617)和纤维杆菌属(P=0.048 ,Q=0.617)序列量显著高于LBF组。
2.6 组间具有显著性差异的分类单元利用LEfSe分析法从门到属的所有分类单元上逐一判断HBF组和LBF组的组间差异,从而筛选关键微生物菌落成员。结果如图5,6所示,HBF组和LBF组间有3个属存在显著性差异,HBF组关键的属为布雷德菌属、梭菌属和考拉杆菌属。
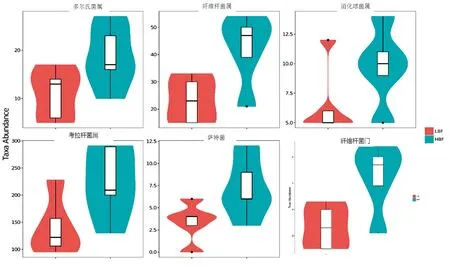
图4 门和属水平组间序列量具显著性差异的分类单元
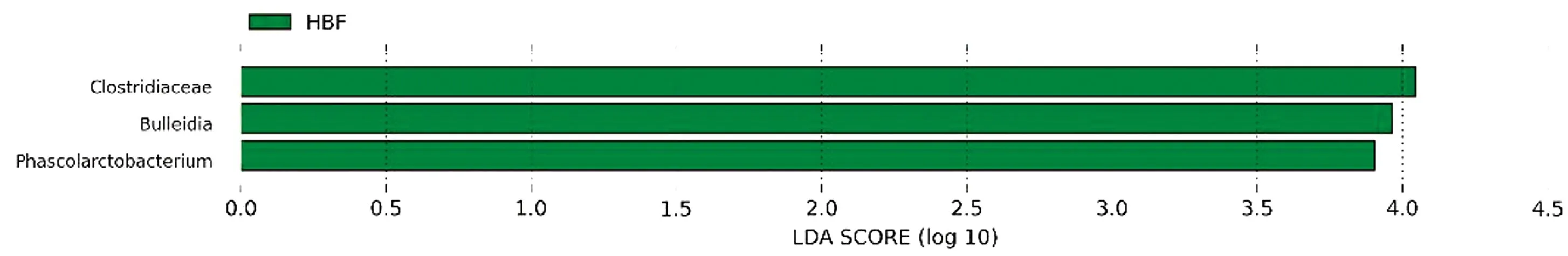
图5 组间具有显著性差异的分类单元
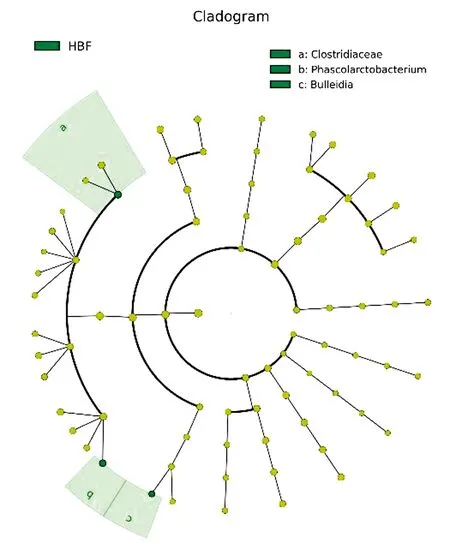
图6 基于分类等级树的组间差异分类单元展示图
2.7 菌群代谢功能预测利用PICRUSt菌群代谢功能预测工具分析了HBF组和LBF组微生物菌群的不同代谢途径,包括代谢、遗传信息处理、环境信息处理、细胞进程、生物体系统和人类疾病。结果显示,LBF组有21条代谢通路较为丰富,其中复制和修复、翻译、氨基酸代谢、聚糖的生物合成和代谢、其他氨基酸的代谢、核苷酸代谢等6条代谢通路的相对丰度高于HBF组;HBF组中有8条代谢通路比LBF组更为丰富,其中与细胞运动性、信号传导的2条通路的相对丰度高于LBF组(图7)。
3 讨论
以往关于猪肠道微生物群对脂肪代谢影响的研究,多选择不同脂肪型遗传背景差别较大的个体来进行,而在同一品种背景下进行的研究有限。在猪的育种过程中为了提高瘦肉率,常把背膘厚作为种公猪的重要经济性状,进行负向选择。我们在军牧1号白猪中通过测定挑选出背膘差异极显著的高背膘和低背膘公猪进行了肠道微生物多样性比较研究,减少了遗传背景差异对研究结果的影响。
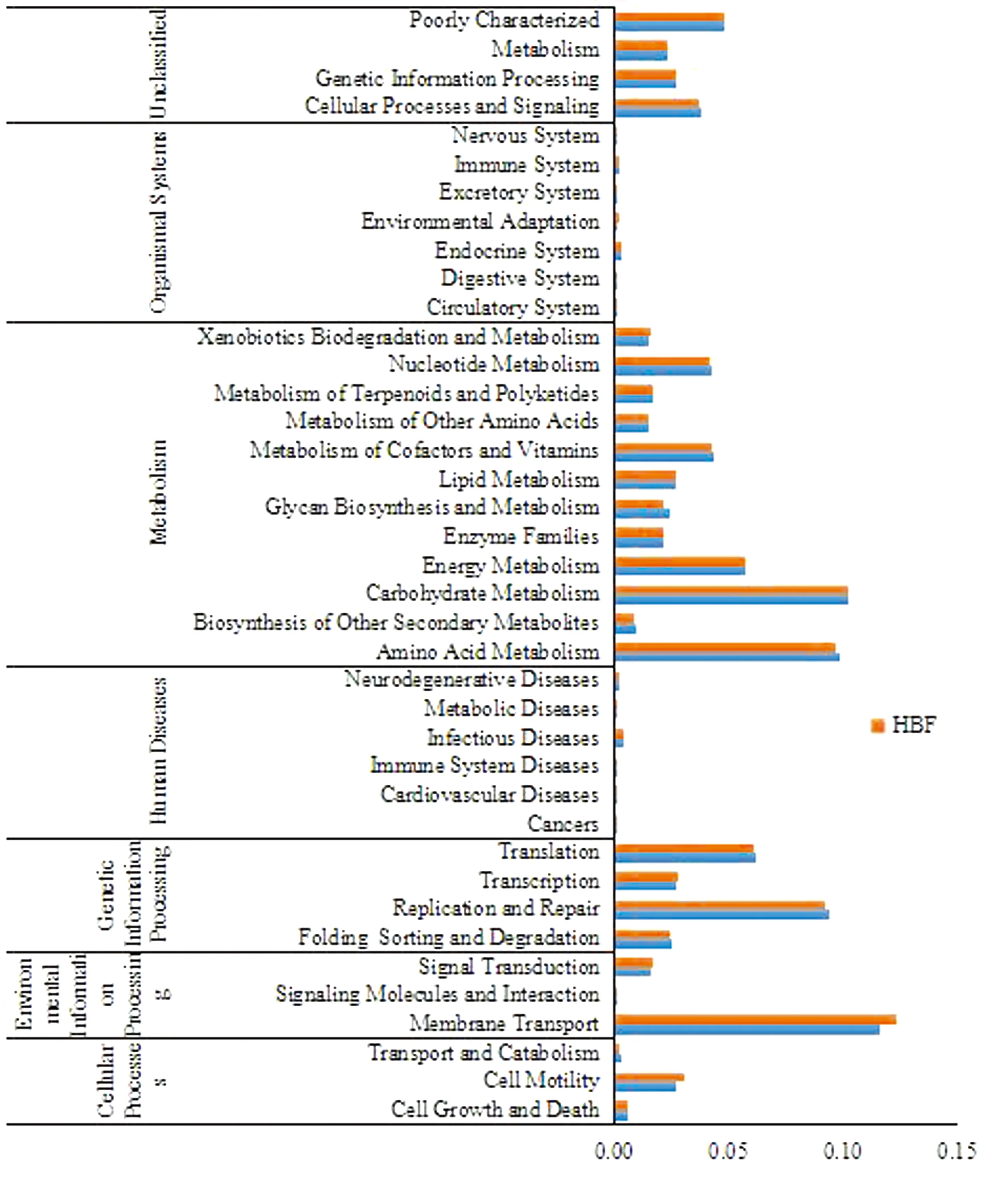
图7 PICRUSt预测的KEGG第二等级分布图
在猪的肠道微生物群中,厚壁菌门和拟杆菌门是2个优势菌门,占总序列的90%以上,其他门如变型菌门、螺旋体门等加在一起平均不到总群落的5%。厚壁菌门和拟杆菌门在不同品种中的组成不同,杜洛克猪(81.7%,P=0.064)、长白猪(83.2%,P=0.030)和大约克夏猪(87.0%,P=0.003)粪便中厚壁菌门所占比例显著高于汉普夏猪(74.2%)。与此相反,汉普夏猪粪便中拟杆菌门的比例(22.2%)高于杜洛克猪(14.8%,P=0.065)、长白猪(12.5%,P=0.019)和约克夏猪(9.13%,P=0.002)[8]。与以往的研究结果相同,军牧1号白猪LBF组和HBF组最丰富的门是厚壁菌门、拟杆菌门、变型菌门和螺旋体门,这4个门在所有样品中的丰度超过90%。
在比较不同脂肪型猪肠道微生物多样性时发现,金华猪的厚壁杆菌门相对丰度(81.57%)高于长白猪(78.70%),而长白猪拟杆菌门的相对丰度(7.95%)高于金华猪(1.15%)[9];荣昌猪(45.39%)和藏猪(39.19%)的厚壁杆菌门均高于大白猪(35.34%),而大白猪(50.83%)的拟杆菌门高于荣昌猪(34.13%)和藏猪(36.31%)[10]。GUO等[11]采用实时PCR对肥胖与瘦八马香猪厚壁菌门和拟杆菌门的相对丰度进行检测,发现脂肪的储存可能影响肠道拟杆菌门的比例。本试验结果表明,在军牧1号中,HBF组厚壁菌门的相对丰度高于LBF组7.55%,而拟杆菌门的相对丰度低于LBF组7.33%,与我国的地方品种较为相似,这可能与军牧1号白猪还有少量的东北民猪的血统有关。同时,结果也证实了在同一品种猪中高背膘的厚壁菌门比例增加,而拟杆菌门的比例下降。
本试验通过Galaxy在线分析平台,提交属水平的相对丰度矩阵进行LEfSe分析,挑取组间具有显著差异的分类单元,结果HBF组仅在厚壁杆菌门的梭菌科、韦荣菌科的考拉杆菌属、厚壁杆菌门丹毒丝菌目丹毒丝菌科的布雷德菌属3个类群的相对丰度均值显著高于LBF组。通过比较不同脂肪型猪肠道微生物多样性发现,脂肪型金华猪盲肠和结肠梭菌科相对丰度显著高于瘦肉型长白猪[8]。TAN等[12]报道,低饲料转化率猪的雷德菌属显著高于高饲料转化率猪。CHRISTINE等[13]研究发现,在给小鼠饲喂高脂肪日粮时,与普通日粮相比,在肠道微生物的组成上厚壁菌门的丹毒丝菌目的比例有所增加。以往关于人或鼠肠道微生物与机体脂肪代谢方面的研究,在筛选关键的生物标记物(即关键群落成员)时所得到的差异显著的分类单元的结果都不尽相同。这可能因为肠道微生物的组成受物种、遗传背景、生长阶段、生理状态、生活环境等多方面因素影响,今后将持续开展不同遗传背景在不同生长阶段、生理状态、生活环境下猪肠道微生物组成变化的研究,以期通过利用肠道微生物群来调控猪肉品质提供一定的策略。
