4例全面发育迟缓伴特征性脑电图异常患儿的临床分析
2020-09-23万瑞平吴本泽刘志刚董诗伟黄小霏
万瑞平 吴本泽 刘志刚 董诗伟 黄小霏

[摘要]目的 分析全面發育迟缓伴特征性脑电图异常患儿的临床特点和病因。方法 对2018年1月~2019年9月在南方医科大学附属佛山妇幼保健院儿童康复科就诊并经视频脑电图检查发现特征性脑电图异常的4例发育迟缓/智力障碍患儿的临床特点进行回顾性分析,总结分析Gesell发育诊断量表、遗传学检查等辅助检查结果。结果 患儿首诊年龄为6~21个月,出生史、家族史均无明确异常。所有患儿均以“发育落后”为首诊原因,Gesell发育诊断量表评估可见各个能区均存在不同程度的发育迟缓。3例皮肤色素减退,2例存在经常发笑、活动过度,均未见癫痫或热性惊厥。所有患儿脑电图均存在特征性表现,其中3例主要表现为前头部为著的节律性高-极高波幅δ波阵发或连续发放,伴后头部节律性棘慢波发放;1例主要表现为广泛性高波幅4~6 Hz θ活动。甲基化特异性多重连接依赖探针扩增检测结果发现3例为15q11-13母源片段缺失,1例存在15q11-13父源单亲二倍体,最后诊断为Angelman综合征。结论 Angelman综合征患儿早期临床表现缺乏特异性或容易被忽略,对于不明原因的发育迟缓/智力障碍患儿,即使没有抽搐表现,仍建议常规进行脑电图检查。Angelman综合征患儿脑电图有一定的特征性,可为早期诊断提供线索,指导进一步遗传学检查以明确诊断。
[关键词]全面发育迟缓;智力障碍;特征性脑电图;Angelman综合征
[中图分类号] R729 [文献标识码] A [文章编号] 1674-4721(2020)6(c)-0009-04
Clinical analysis of 4 cases of children with global developmental delay and characteristic electroencephalogram pattern
WAN Rui-ping1 WU Ben-ze2 LIU Zhi-gang2 DONG Shi-wei2 HUANG Xiao-fei1
1. Department of Children Rehabilitation, Foshan Maternity and Child Healthcare Hospital Affiliated to Southern Medical University, Guangdong Province, Foshan 528000, China; 2. Department of Pediatric, Foshan Maternity and Child Healthcare Hospital Affiliated to Nanfang Medical University, Guangdong Province, Foshan 528000, China
[Abstract] Objective To analyze the clinical characteristics and etiology of children with comprehensive developmental delay and characteristic EEG abnormalities. Methods From January 2018 to September 2019, 4 cases of children with developmental retardation/mental disability were diagnosed by video EEG examination in the Department of Pediatric Rehabilitation, Foshan Maternal and Child Health Hospital Affiliated to Southern Medical University. The characteristics were retrospectively analyzed, and the results of auxiliary examinations such as Gesell Development Diagnostic Scale and genetic examination were summarized and analyzed. Results The children′s age at the first diagnosis was 6-21 months, and there were no definite abnormalities in the birth history and family history. All children were diagnosed with "developmental backwardness" as the first reason. Gesell′s Developmental Diagnostic Scale showed that there were various degrees of developmental delay in each energy zone. Three children had hypopigmentation of the skin, and 2 cases had frequent laughter and hyperactivity, and neither had epilepsy or febrile seizures. All children′s electroencephalograms had characteristic manifestations. Among them, 3 cases mainly showed that the front head was markedly rhythmic high-very high amplitude delta wave burst or continuous distribution, with posterior head rhythmic spike slow wave distribution. One case mainly exhibited extensive high-amplitude 4-6 Hz θ activity. Methylation-specific multiple ligation-dependent probe amplification detection results showed that 3 children had 15q11-13 maternal fragments missing, 1 patient had 15q11-13 paternal diploid, and finally diagnosed Angelman syndrome. Conclusion The early clinical manifestations of children with Angelman syndrome lack specificity or are easily overlooked. For children with unexplained stunting/mental disability, even if there is no convulsions, routine EEG examination is recommended. The EEG of children with Angelman syndrome has certain characteristics, which can provide clues for early diagnosis and guide further genetic examination to confirm the diagnosis.
[Key words] Global developmental delay; Intellectual disability; Characteristic electroencephalogram pattern; Angelman syndrome
全面发育迟缓(global developmental delay,GDD)是指5岁以下处于发育早期的儿童,存在运动、语言、认知、社交、日常生活能力等2个及以上能区的发育落后[1-2]。GDD患儿5岁以后通常会发展成为智力障碍(intellectual disability,ID),故临床上一般统称为GDD/ID[3-4]。由于GDD/ID存在多种多样的病因,大多数患儿在脑电图(electroencephalogram,EEG)上并没有特异性的表现[5]。Angelman综合征(Angelman syndrome,AS)是以15q11-13染色体区域基因异常引起的遗传性神经发育障碍性疾病。AS患儿均存在发育落后,早期以运动、语言发育迟缓就诊,临床表现缺乏特异性,导致诊断有一定的困难[6-7]。既往研究表明,AS患儿具有相对特征性的EEG异常,可为早期诊断AS提供线索[8-10]。然而,目前仍有不少医技人员缺乏对这种特征性EEG的重视。本研究通过分析4例伴有特征性EEG异常的GDD/ID患儿的临床诊疗过程,希望促进对该方面的认识,从而帮助GDD/ID患儿的病因诊断。
1資料与方法
1.1一般资料
以2018年1月~2019年9月在南方医科大学附属佛山妇幼保健院儿童康复科就诊,并进行视频EEG检查发现特征性EEG异常的4例GDD/ID患儿为研究对象。特征性EEG异常包括:①δ图形:前头部为著的节律性高-极高波幅(200~500 μV)2~3 Hz δ活动或三相δ波;②θ图形:持续高波幅(≤200 μV)广泛性或后头部4~6 Hz θ活动;③后头部棘慢波图形:后头部节律性棘慢波混合高波幅(>200 μV)3~4 Hz慢波活动[9]。所有患儿最后均经遗传学检查确诊为AS。
1.2方法
1.2.1临床资料收集 详细收集患儿的临床资料,包括母孕史、出生史、生长发育史、既住史、家族史等病史,头围、皮肤和毛发、面容特征、神经系统查体等体征,生化检查、血尿遗传代谢筛查、头颅影像学、Gesell发育诊断量表评估等检查结果。Gesell发育诊断量表评分标准如下:75~84分为边缘水平,55~74分为轻度低下,40~54分为中度低下,25~39分为重度低下,<25分为极重度低下。
1.2.2视频EEG检查 采用32导联视频EEG监测,记录包括清醒和入睡的至少一个完整睡眠周期。结果由儿童神经专科医师及电生理技师共同判读。
1.2.3遗传学检查 经患儿家长签字同意,抽取患儿外周静脉血提取DNA。采用甲基化特异性多重连接依赖探针扩增(methylation-specific multiplex ligation-dependent probe amplification,MS-MLPA)技术检测染色体15q11-13区域变异。采用二代测序技术检测基因全外显子变异。
2结果
2.1临床资料分析
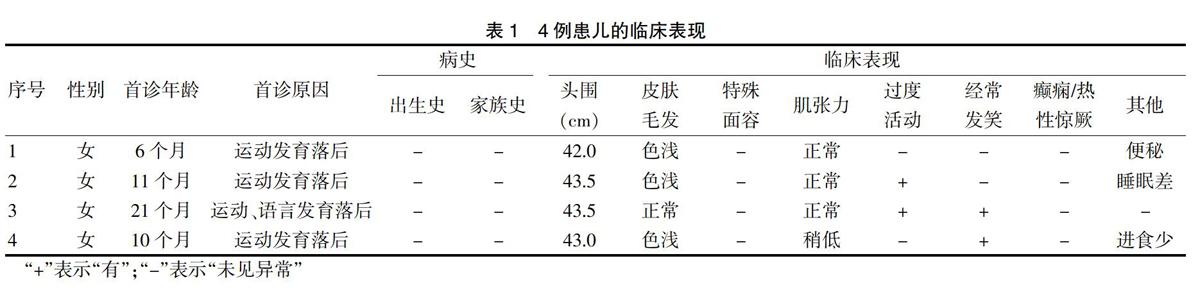
4例患儿均为女孩,首次就诊年龄为6~21个月。所有患儿的出生史、家族史病史均无明确异常,无发育迟缓、抽搐家族史。1例出生后第2天因新生儿高胆红素血症住院3 d,1例母亲怀孕早期有带状疱疹病毒感染。所有患儿均以“发育落后”为首诊原因,Gesell发育诊断量表评估可见患儿各个能区均存在不同程度的发育迟缓。3例皮肤和毛皮颜色较浅,2例存在经常发笑行为,2例存在活动过度表现,1例便秘,1例睡眠差和进食少。首诊时所有患儿均未见癫痫或热性惊厥(表1)。
2.2视频EEG检查结果分析
所有患儿EEG均见特征性表现,其中3例EEG主要表现为前头部为著的节律性高-极高波幅δ波阵发或连续发放,伴后头部节律性棘慢波发放;1例主要表现为广泛性高波幅4~6 Hz θ活动。头颅MRI方面,可见脑外间隙增宽或白质髓鞘化延迟/未完善等非特异性表现。血尿遗传代谢筛查均未见异常(表2)。
2.3遗传学检查结果分析
4例患儿均行MS-MLPA检测,结果发现3例患儿存在15q11-13母源片段缺失,1例患儿存在15q11-13父源单亲二倍体。1例患儿在MS-MLPA检测之前曾行二代测序技术检测基因全外显子,结果未检测到致病性变异(表2)。
2.4随访结果
随访时间为6个月~2年,随访结束时,所有患儿均存在明显的认知障碍,尚未有语言表达。2例患儿出现抽搐发作,其中1例为19个月开始出现发热惊厥,23个月出现无热惊厥,表现为强直-阵挛发作,服用丙戊酸钠(德巴金口服液,赛诺菲,国药准字H20041435)之后可控制发作;另外1例24个月开始出现肌阵挛样发作,但家长未带患儿就诊。
3讨论
GDD/ID是儿童时期最常见的神经发育障碍疾病之一,患病率约为3%[1-2],严重影响患儿的身心健康和生活质量。该病病因复杂,既有外在的环境因素,也有内在的遗传因素。据报道目前仍有50%以上GDD/ID患儿的病因不明,而遗传因素在该部分患儿的发病中起着重要作用[11-12]。AS是由于母源染色体15q11-13上编码泛素蛋白连接酶E3A(UBE3A)基因缺失或表达异常引起的遗传性GDD/ID。国外报道新生儿AS发病率约为1∶50 000~1∶25 000[13-14],我国尚无相关流行病学调查报告。
Williams等[8]在2006年修订了AS的诊断标准,指出100%患儿可出现严重的发育迟缓、运动或平衡障碍(共济失调、肢体震颤)、特殊行为(不合场合的大笑或微笑、易兴奋、拍手、多动)以及语言障碍;超过80%患儿在2岁时出现获得性小头,3岁前出现癫痫发作,2岁前出现特征性的异常EEG;20%~80%患儿可出现吐舌、吸吮或吞咽障碍、婴儿期喂养困难、频繁流涎、过度咀嚼动作、皮肤色素减退、下肢过度活动、睡眠障碍、脊柱侧凸、便秘等相关表现。杨志仙等[9]认为,上述标准为AS提供了概括性的临床诊断依据,但由于AS为罕见疾病,临床症状多样,即便专科医生也常常会因忽略本病的症状和体征而造成漏诊。而且AS患儿各种阳性体征的发生率在≤2岁年龄组明显低于>2岁组[15]。马秀伟等[10]研究显示,AS患儿在婴儿期内最主要的表现是发育迟缓,只有50%存在癫痫发作、与环境不相适应的爱笑,未发现明显共济失调。本文4例患儿首诊年龄<2岁,就诊时均存在发育迟缓,3例患儿皮肤色素减退,2例存在经常发笑、活动过度,均未见癫痫或热性惊厥。由上可见,AS患儿早期临床表现缺乏特异性或容易被忽略,年龄越小越不容易被诊断。
AS患儿具有较为特征性的EEG,包括了前头部为著的高-极高波幅2~3 Hz δ活动、广泛性或后头部高波幅4~6 Hz θ活动、后头部棘慢波混合高波幅3~4 Hz慢波活动[9,16]。这些特征性的EEG改变,对临床早期诊断工作提供了很大帮助。既往研究表明,AS患儿EEG改变出现较早,通常出现在2岁之前,早于临床表现出现,而且与临床癫痫发作无明显相关性,可为早期诊断AS提供线索[17]。国内多个学者在研究中指出,多数AS病例的早期诊断得益于EEG特征性异常的提示[9-10]。本研究中4例患儿也是在EEG检查发现特征性EEG异常,然后进行遗传学检查确诊AS。因此,对于不明原因的GDD/ID患儿,特别是中重度障碍患儿,即使没有抽搐表现,仍建议常规进行EEG检查以帮助提示病因诊断。但需要注意的是,AS患儿EEG的特征性并不等同于特异性,上述3种EEG图形也可见于其他GDD/ID或癫痫的患儿,需要结合其他临床特点进行鉴别。此外,有少部分AS患儿并不是一开始就出现特征性EEG改变,需要进行多次检测,即使EEG阴性也不能排除AS诊断,必须综合分析病情特点,必要时行遗传学检查。
AS可由4种不同方式的基因异常所致,其中母源15q11-13缺失约占70%、父源单亲二倍体占2%~7%、印记中心缺陷占3%~5%、UBE3A基因突变约占10%[6,18]。遗传学检查是确诊AS的主要方法,MS-MLPA、甲基化PCR是目前最为常用的检测方法,可以检出70%~80%的AS患者。对于上述2种方法检测阴性,但临床高度怀疑AS者,应进一步行基因测序以明确是否存在UBE3A基因点突变或小片段缺失。本文4例患儿均行MS-MLPA检测,3例为15q11-13母源片段缺失,1例为15q11-13父源单亲二倍体。值得提出的是,1例在MS-MLPA检测之前曾行二代测序,结果未检测到致病性变异。鉴于目前多数基因公司的二代测序难以准确检测大片段缺失,而该变异类型为AS主要致病变异,二代测序可能导致漏筛。因此,临床上应先综合分析患儿临床表现和EEG特征等指标,再选择适合的遗传检查方法以提高检测阳性率。
综上所述,AS患儿早期临床表现缺乏特异性或容易被忽略,对于不明原因的GDD/ID患儿,即使没有抽搐表现,仍建议进行常规EEG检查。AS患儿EEG有一定的特征性,可为早期诊断提供线索,指导进一步遗传学检查以明确诊断。
[参考文献]
[1]Moeschler JB,Shevell M,Committee on Genetics.Comprehensive evaluation of the child with intellectual disability or global developmental delays[J].Pediatrics,2014,134(3):e903-e918.
[2]中华医学会儿科学分会神经学组,中国医师协会神经内科分会儿童神经疾病专业委员会.儿童智力障碍或全面发育迟缓病因诊断策略专家共识[J].中华儿科杂志,2018, 56(11):806-810.
[3]Purugganan O.Intellectual Disabilities[J].Pediatr Rev,2018, 39(6):299-309.
[4]Belanger SA,Caron J.Evaluation of the child with global developmental delay and intellectual disability[J].Paediatr Child Health,2018,23(6):403-419.
[5]劉晓燕.小儿神经系统某些常见病的EEG表现[J].临床神经电生理学杂志,2007,16(2):112-115.
[6]刘依竞,肖农.Angelman综合征发病机制、分型及治疗进展[J].临床儿科杂志,2015,33(7):668-672
[7]邓亚萍,曹洁.15号染色体相关性癫痫综合征诊疗进展[J].临床儿科杂志,2015,12(33):1073-1076.
[8]Williams CA,Beaudet AL,Clayton-Smith J,et al.Angelman syndrome 2005: updated consensus for diagnostic criteria[J].Am J Med Genet A,2006,140(5):413-418.
[9]杨志仙,刘晓燕,秦炯,等.Angelman综合征临床及脑电图特征[J].中国实用儿科杂志,2011,26(7):519-523.
[10]马秀伟,温秀芳,刘京京,等.婴儿期诊断Angelman综合征5例分析并文献复习[J].中国儿童保健杂志,2015,23(4):446-448.
[11]杨璞,桂宝恒,邬玲仟.智力障碍的病因及诊断方法[J].中国当代儿科杂志,2015,17(6):543-548.
[12]Vasudevan P,Suri M.A clinical approach to developmental delay and intellectual disability[J].Clin Med(Lond),2017, 17(6):558-561.
[13]Oiglane-Shlik E,Talvik T,Zordania R,et al.Prevalence of Angelman syndrome and Prader-Willi syndrome in Estonian children: sister syndromes not equally represented[J].Am J Med Genet A,2006,140(18):1936-1943.
[14]Mertz LG,Christensen R,Vogel I,et al.Angelman syndrome in Denmark birth incidence,genetic findings,and age at diagnosis[J].Am J Med Genet A,2013,161A(9):2197-2203.
[15]白晋丽,宋昉,邹丽萍,等.15q11-13缺失型Angelman综合征17例遗传学诊断及临床特点[J].中华儿科杂志,2010, 48(12):939-943.
[16]孔令宇,廖建湘.Angelman综合征脑电图特点的研究进展[J].国际儿科学杂志,2017,44(11):780-782.
[17]Laan LA,Vein AA.Angelman syndrome is there a characteristic EEG[J].Brain Dev,2005,27:80-87.
[18]Sadikovic B,Fernandes P,Zhang VW,et al.Mutation update for UBE3A variants in Angelman syndrome[J].Hum Mutat,2014,35(12):1407-1417.
(收稿日期:2019-12-31 本文编辑:刘振宇)
[基金项目]广东省佛山市科技局医学类科技攻关项目(2017 AB002 981)
[作者简介]万瑞平(1982-),女,广东佛山人,医学博士,主治医师,主要从事儿童神经康复、儿童神经遗传性疾病研究
通讯作者:刘志刚,主任医师
