三阴性乳腺癌肿瘤微环境特征免疫相关生物学标志物筛选及功能预测分析
2020-09-15苏芃毛晓韵关舒崔梦遥金紫凝金锋
苏芃, 毛晓韵, 关舒, 崔梦遥, 金紫凝, 金锋
三阴性乳腺癌(triple-negative breast cancer,TNBC)是乳腺癌的特殊亚型,其特征为缺乏雌激素受体(ER)和孕激素受体(PgR)的表达,以及缺乏人表皮生长因子受体2(HER2)的表达。一直以来TNBC由于其具有高侵袭性、高转移性和显著的耐药性而备受关注[1]。又因缺少有效的治疗靶点,TNBC患者的数量虽然只占乳腺癌患者总数的20%[2],但是死亡人数却占所有乳腺癌患者死亡人数的80%[3]。研究表明,大多数TNBC患者发生肺转移和骨转移后,中位总生存时间(overall survival, OS)为12~18个月[4]。随着全球TNBC患者不断增加,寻找有效的治疗靶点和预后生物学标志物成为TNBC临床和科研的重点及难点。
目前手术和放化疗仍是治疗TNBC的常规方式。目前批准的化疗药物包括蒽环类、紫杉类和铂类等[5-7],但是由于TNBC的致癌因子和耐药性的异质性,这些化疗药物的疗效非常有限[8-9]。既往研究表明,在其他实体性肿瘤中,患者可以通过免疫治疗获益。免疫检查点抑制剂(immune checkpoint inhibitors,ICIs)已经被证明是目前最有效的免疫治疗药物,其能阻断免疫抑制受体(CTL-4和PD-1)提高肿瘤浸润淋巴细胞(TILs)的细胞毒性和增殖能力[10-17]。相比其他乳腺癌亚型,TNBC被认为是最有可能从免疫治疗中获益的。首先,TNBC中存在大量的TILs,而TILs在其他肿瘤中已经被证明与ICIs疗效呈正相关;其次,在TNBC中PD-L1的表达在肿瘤组织和免疫细胞中均高表达,这为ICIs提供了直接的治疗靶标;再次,TNBC中存在大量的非同义突变,它们会产生肿瘤异质性新抗原,从而激活抗原异质性T细胞,进而产生抗肿瘤免疫应答。但是目前还没有一种有效的治疗TNBC的免疫治疗方案。
肿瘤微环境(tumor microenvironment,TME)已经被证明是肿瘤潜在治疗靶点的重要来源之一,其有很高的复杂性,且越来越多的文献表明,TME在肿瘤进展和治疗反应中起着关键作用[18-19]。例如,在诊断时可以通过TME的细胞成分预测免疫治疗疗效[20-21]和化疗受益[22]。已经证明,TME中的CD8+T细胞、CD4+T细胞、巨噬细胞和癌相关成纤维细胞数量的变化与乳腺癌患者的临床预后密切相关[23]。所以如何正确理解TNBC患者的TME是免疫治疗TNBC的重中之重。
本研究利用生物信息学分析方法评价TNBC患者中的免疫微环境细胞浸润情况,希望从免疫微环境层面对TNBC患者的预后进行分析,进而提出新的TNBC免疫预后相关生物学标志物并描述其在TNBC中的生物学过程,为临床治疗TNBC提供新的治疗靶点。
1 资料与方法
1.1 数据下载与标准化 系统检索TNBC相关公开数据库并筛选样本,筛选标准:①数据集包含mRNA表达及生存数据;②TNBC患者样本数>40;③患者总生存时间>10 d;④术前未接受过抗肿瘤治疗。芯片测序数据通过RMA算法进行标准化,对于一个基因对应多个探针的情况,取表达量的中位数作为该基因的表达量,并将基因表达中位数为0标准差小于0.1的基因剔除。
1.2 CIBERSORT 通过CIBERSORT算法[24]评价TNBC中免疫细胞成分包括:B细胞、T细胞、自然杀伤细胞、巨噬细胞、树突状细胞和髓细胞亚群等22种免疫细胞。该算法通过CIBERSORT官方网站(http://cibersort.stanford.edu/)实现,提取P<0.05的分析结果。
1.3 一致性聚类分析 采用无监督聚类法(K-means)对TNBC患者的免疫细胞成分进行聚类,寻找最佳的分类类型。此算法通过R包(Consensu Cluster Plus)实现[25],并通过1 000次交叉验证证明数据结果的可靠性。
1.4 基因差异分析 采用差异基因分析探索TNBC中表达量异常的基因。通过R包(limma)实现,并对基因表达量进行标准化。截断值的选择为|log2foldChange(FC)|≥1并且调整后P<0.05,即TNBC中基因表达量超过癌旁组织中表达量的2倍或降低2倍,统计学有差异的基因即为差异基因。
1.5 基因富集分析 为了计算单样本基因集富集度,我们使用GSEA分析[26-27]推导出先前实验验证的基因特征的绝对富集分数,通过R包(Cluster Profiler)[28]进行GO富集分析和KEGG通路分析,提示基因集主要富集的通路,注释其参与的生物学过程。
1.6 Lasso-Logistic及ROC验证 为了准确筛选免疫亚型的生物标志物,我们通过lasso-logistic模型[29]进行降维筛选。lasso-logistic回归模型的目标函数如下:
其中,λ表示惩罚系数,可以通过10折交叉验证选取最优λ,||α||1定义为每个向量元素绝对值之和。lasso-logistic模型通过R包(glmnet)实现。受试者工作特征曲线(ROC)用来评价降维后数据与免疫亚型之间的关系,ROC曲线下面积(AUC)越大,说明通过降维后数据分类的结果越好。
1.7 网络加权共表达分析(WGCNA) 网络加权共表达分析是利用分子间的表达相关系数来衡量它们的共表达关系,同一模块中的分子表达模式相似,而和其他模块分子表达模式差别较大。WGCNA中使用的方法就是使用dissimilarity来进行聚类,其采用的具体算法是拓扑重叠(topological overlap dissimilarity measure,TOM)以计算基因间的关联程度[30]。
1.8 统计学方法 本研究运用R软件进行统计分析,服从正态分布的定量数据以均数±标准差来表示,组间比较采用t检验,不服从正态分布的定量数据以中位数及四分位间距来表示,组间比较采用Wilcoxon检验。采用Kaplan-Meier法生成各数据集中各亚组的生存曲线,采用Log-rank检验确定差异的统计显著性,P<0.05为有统计学意义。
2 结果
2.1 研究对象特征 检索GEO公开数据库,下载GSE103091数据,通过整理共纳入包含mRNA数据和完整总生存数据的TNBC患者100例。患者平均年龄为(57.0±13.1)岁;生存72例,死亡28例,中位生存时间为66.47个月;70例患者未转移,30例(30%)出现远处转移。
2.2 一致性聚类分析(Consensus Cluster) 为选择最合适的分组,本研究通过R包(Consensus Cluster Plus)进行,通过一致性聚类分析结果发现,TNBC患者免疫细胞成分可分为两类,一类为低免疫浸润组,一类为高免疫浸润组。低免疫浸润组患者43例,其中转移患者19例(44.2%);高免疫浸润组患者57例,其中转移患者11例(19.3%),两组转移患者比例差异有统计学意义(χ2=6.09,P=0.01),见图1。通过生存分析发现低免疫浸润组患者总生存较低,预后不良,见图2。通过GSEA分析发现低免疫浸润组人群主要受到正向免疫调节相关通路和细胞刺激因子相关通路的调控,见图3,表1。

表1 免疫低浸润组功能富集分析

图1 三阴性乳腺癌免疫成分的一致性聚类分析

图2 免疫分组相关生存分析
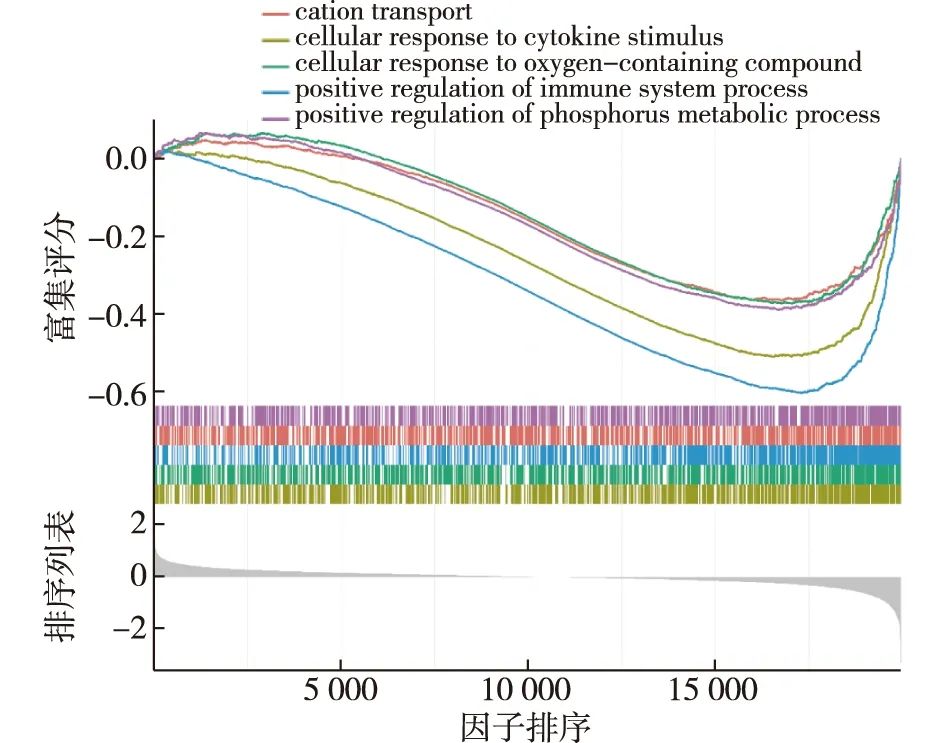
图3 低免疫浸润组的功能富集分析
2.3 基因差异分析 通过R包(limma)对TNBC患者的癌与癌旁组织样本进行基因差异分析,得到1 304个差异基因,其中422个为高表达差异基因,882个为低表达差异基因。
2.4 筛选免疫分组的生物学标志物 为了筛选上述免疫分组的生物学标志物,我们通过1 000次Lasso-Logistic对TNBC差异分析基因进行降维分析。结果发现有36个基因可以作为免疫分组的生物学标志物(图4),通过单因素Cox分析筛选与TNBC患者总生存相关的基因1 726个。上述结果相互取交集,得到36个免疫分组生物学标志物中有5个基因与TNBC患者总生存相关,见图5。通过ROC验证此5个基因可以有效地反映免疫分组情况,AUC面积为0.946,见图6。

4A:1 000次lasso logistic中每次分析的基因组合;4B:1 000次分析中各个基因出现的次数,黑色虚线为出现900次,黑色虚线以上为该基因出现过900次以上图4 免疫分型生物学标志物筛选

图5 免疫分型核心基因筛选

图6 核心基因的ROC曲线分析
2.5 免疫分组生物学标志物的生物功能分析 为了描述上述分析得到的5个免疫分组生物学标志物(FOLH1、WDR18、LINC00638、OAS3、SETDB2),我们采用WGCNA进行共表达分析,再通过基因富集分析描述与此5个基因共表达模块的生物学功能,推测此5个基因的生物学功能。结果发现,FOLH1共表达在黑色模块中,WDR18共表达在天青色模块中,这两个基因主要生物学功能与肿瘤增殖相关;LINC00638共表达在棕色模块中,主要生物学功能与T细胞激活等免疫相关;OAS3共表达在黄绿色模块中,主要生物学功能与病毒基因降解通路相关;SETDB2被分类在灰色模块中,由于灰色模块为非聚类基因集合,所以SETDB2在TNBC中发挥的作用还需要进一步研究。见图7。
3 讨论
肿瘤微环境的改变已经被广泛认为是可以影响TNBC患者预后的重要靶标。但是我们发现目前对于肿瘤微环境与TNBC的研究略有不足,而且还没有通过免疫微环境细胞成分分析TNBC患者预后的研究。本研究对TNBC患者的免疫微环境细胞浸润情况进行分析,提出5个免疫相关预后生物学标志物,并描述了其在TNBC患者中的生物功能,为TNBC免疫治疗提供了新的靶点。
有研究已经证实,TNBC并不是传统认识中的单一类型,同为TNBC亚型的患者存在着较大的生存差异,对不同治疗方案的敏感性也不同。这与本研究的结论相同。本研究通过Cibersort对TNBC患者的TME细胞成分进行分析,并且发现TNBC患者可以通过TME细胞成分分为两组,即“高免疫浸润组”和“低免疫浸润组”。结果显示,低免疫浸润组的人群预后不良,并且通过GSEA分析发现低免疫浸润组人群中正向免疫调节相关通路和细胞刺激因子相关通路发挥了重要的作用。通过1 000次logisticlasso回归进行特征选取,最后筛选出5个基因(FOLH1、WDR18、LINC00638、OAS3、SETDB2)为免疫相关预后标志物,再通过WGCNA分析发现,WDR18在TNBC参与mRNA编辑过程影响预后。有研究表明,WDR18可以与TopBP1共同促进DNA损伤检查点信号传导[31],DNA损伤已经被证明在乳腺癌中发挥重要的作用,与本研究的结果一致。还有研究发现,LINC00638基因可以与HCP5、XIST和TP53TG1在新生儿败血症中通过内源性RNA作用影响患儿预后[32]。LncRNA作为非编码RNA本身没有编码蛋白的功能,但是其可以调节mRNA,是非常重要的。本研究结果发现LINC00638是TNBC免疫相关预后生物学标志物,参与T细胞激活等重要免疫通路,目前对于LINC00638的研究不足,还需要进一步研究。据报道,SETDB2是1型IFN信号传导下游的干扰素刺激基因(ISG),并负责减弱Ⅰ型IFN和转录因子NFkB诱导的促炎和抗病毒基因[33-34];SETDB2在肿瘤中的异常表达与患者的耐药性有关[35];SETDB2的低表达与晚期肾细胞肿瘤的转移扩散有关[36]。本研究发现,在TNBC患者中SETDB2高表达患者的预后比SETDB2低表达患者的预后好,这说明在TNBC中SETDB2是一个重要的预后保护因素,但是SETDB2在TNBC中发挥的生物学功能还需要进一步的研究。OAS3作为经典的干扰素靶基因,已经被证实参与细胞凋亡过程[37],并且与宫颈癌HPV感染相关且影响患者预后,迄今为止还没有报道其与乳腺癌相关。本研究发现,OAS3在TNBC患者中参与病毒基因降解功能,其低表达患者预后不良,为TNBC的预后保护因素。

7A:TNBC免疫相关基因的共表达聚类分析;7B:FOLH1生物学功能预测;7C:WDR18生物学功能预测;7D:LINC00638生物学功能预测;7E:OA53生物学功能预测图7 WGCNA及核心基因的生物学功能预测
综上所述,本研究筛选出TNBC患者免疫相关预后生物学标志FOLH1、WDR18、LINC00638、OAS3及SETDB2,并通过验证它们可以反映TNBC患者的预后情况。但本研究还有一些缺点与不足:①研究的样本量较小,对于TNBC总体人群的代表性有限,还需要后续大样本的研究;②提出的生物学标志物还需要分子细胞学实验的证明,分子机制还需要进一步的讨论;③采用芯片测序技术,由于技术本身的限制,存在批次效应有可能会导致数据的偏倚;④本研究分析结果还需要更多数据集进行验证。
