层黏连蛋白α2缺陷型先天性肌营养不良的临床与基因分析
2020-08-24何展文李平甘吴若豪陈启慧罗向阳
何展文 柏 萍 李平甘 吴若豪 陈启慧 李 宇 罗向阳
中山大学孙逸仙纪念医院,广东 广州 510120
先天性肌营养不良(congenital muscular dystrophy,CMD)是一组自出生时或生后数月内起病的罕见的原发性和进行性肌病,具有临床和遗传异质性,主要临床表现为婴儿早期出现肌无力、运动发育迟缓、肌张力低下等[1]。层黏连蛋白α2缺陷型先天性肌营养不良(LAMA2-MD)是CMD最常见的亚型,占所有CMD的1/3,LAMA2-MD与编码层黏连蛋白2(laminin2)的3个亚单位之一α2链(LAMA2,也称Merosin)基因缺陷有关[2-3]。肌肉活检及LAMA2基因检测可明确其诊断,但至今国内基因明确病例并不多见。本研究回顾2010-10—2018-09中山大学孙逸仙纪念医院确诊3例LAMA2-MD的临床特点和基因结果,探讨临床表现以及基因检测对诊断的作用。
1 资料与方法
1.1诊断标准LAMA2-MD诊断主要根据临床表现和实验室检查,主要临床表现包括出生时至婴儿早期出现肌无力、肌张力减低,运动发育落后,关节挛缩,智力多正常,血清肌酸激酶水平增高。确诊经肌肉活检病理和基因检测,肌肉病理免疫组化染色提示Merosin完全缺失或部分表达,或基因检测到两个致病性等位LAMA2基因突变[3]。
1.2研究方法
1.2.1 头颅影像学检查:采用Philips Intera 1.5 T超导MR仪,仰卧位,行头颅平扫和增强检查,常规SE序列T1WI(TR 450 ms,TE 15 ms)成像,T2WI(TR 2 000 ms,TE 120 ms)成像,层厚5 mm,间隔0.5 mm,矩阵256×256,FOV 230 mm×230 mm,采用Gd-DTPA(剂量0.2 mL/kg体质量)对比剂,分别行横轴位和冠状位、矢状位检查。
1.2.2 肌肉病理:首先向患儿家属交代病情和手术相关事项,签署同意书。选择小腿腓肠肌行开放性活检。肌肉标本分成两部分,一部分经异戊烷预冷后放入液氮中固定,冰冻切片,切片厚度8 μm。分别进行组织学和组织化学方法染色,包括苏木精-伊红(HE)染色、改良 Gomori三色(MGT)染色、还原型辅酶Ⅰ四氮唑还原酶(NADH-TR)染色、高碘酸 Schiff反应(PAS)染色、油红O染色、琥珀酸脱氢酶(SDH)染色、细胞色素C氧化酶(COX)和染色SDH/COX双染。另一部分肌肉在2%戊二醛中固定,1%锇酸后固定,常规脱水和塑料包埋,超薄切片,铅铀双染色,电镜下观察。
1.2.3 肌肉标本免疫组化:单克隆一抗包括抗萎缩蛋白(Dystrophin)-Rod、Dystrophin-C、Dystrophin-N、Dysferlin、α/β/γ/δ-肌聚糖(sarcoglycan)、层黏连蛋白(Merosin)及胶原蛋白Ⅵ(Collagen Ⅵ)抗体,免疫组织化学染色采用链霉卵白素-生物素法染色试剂盒。
1.2.4 致病基因检测:收集患儿及父母外周血标本,提取DNA,二代基因测序检测遗传性肌营养不良基因,多重链接探针扩增技术(MLPA)检测LAMA2基因缺失或重复。
2 结果
2.1临床表现确诊层黏连蛋白α2链缺陷型先天性肌营养不良患儿共3例(表1),男2例,女1例。所有患儿均为生后起病,表现为运动里程碑落后,近端肌无力为主,肌张力低下。例2能独走,但走路不稳,上楼梯困难,其余2例患儿最大运动能力为独坐。语言发育与智力均正常。3例患儿营养状态不良,体质量低下。例1有脊柱侧弯,例1和例3有关节挛缩(肩、肘、膝和踝关节)。血清肌酸激酶(CK)中度升高(895~1 414 U/L),肌酸激酶同工酶(CK-MB)轻中度升高(34~94 U/L)。所有患儿行心电图检查均无异常。

表1 3例层黏连蛋白α2链缺陷型先天性肌营养不良患儿的临床特点Table 1 Clinical characteristics in 3 patients with LAMA2-MD
2.2脑电图与头颅磁共振检查所有患儿行脑电图及头颅MRI检查。3例患儿脑电图检查示正常小儿脑电图。3例患儿头颅MRI检查均示双侧大脑半球白质区对称多发斑片状、片状异常信号灶,T1WI上呈稍低信号(图1A),T2WI上呈高信号(图1B),FLAIR上呈高信号(图1C),增强未见明显强化(图1D);内囊、胼胝体、基底节、丘脑、小脑及脑干均无受累,侧脑室、第三、第四脑室形态、信号未见异常。
2.3肌电图、肌肉病理及免疫组织化学检查全部病例行肌电图检查,肌电图检查均提示肌源性损害,表现为周围神经运动传导波幅低,时限缩短。病例2和病例3进行肌肉组织病理检查。肌肉病理组织学HE染色显示肌纤维大小不等,可见散在分布少许坏死伴吞噬纤维及再生纤维;可见较多高收缩纤维,未见内核纤维增多;肌间质未见炎细胞浸润,肌间及肌内膜结缔组织明显增生(图2A)。电镜检查示部分肌纤维内肌原纤维排列紊乱,局部见灶状肌溶灶及萎缩肌纤维,局部脂质空泡增多、糖原未见增多,未见杆状体改变,未见异常管聚集现象;未见异常线粒体堆积,肌间质胶原纤维增生,未见炎细胞浸润,毛细血管基底膜未见明显增厚(图2B)。病例2免疫组织化学染色显示Merosin部分肌纤维膜表达减弱,局灶缺失(图2C);病例3免疫组织化学染色显示Merosin表达明显减弱或缺失(图2D)。Dystrophin-Rod/C/N、Dysferlin、Collagen Ⅵ和α/β/γ/δ-sarcoglycan均完全正常,表达连续、均匀。
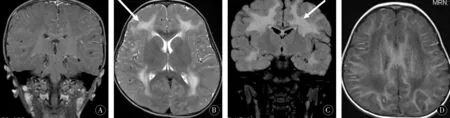
图1 层黏连蛋白α2链缺陷型先天性肌营养不良患儿头颅MRI表现,脑实质结构对称,脑室不大,双侧大脑半球白质信号异常,A:T1WI上呈稍低信号(冠状位);B:T2WI上呈高信号(横断位);C:FLAIR上呈高信号(冠状位);D:增强未见明显强化(横断位)Figure 1 Skull MRI manifestations in patients with LAMA2-MD.The parenchymal structure of the brain is symmetrical,the ventricles are small,and the white matter signals of the cerebral hemispheres are abnormal.A:Slightly low signal on T1WI (Coronal position);B:High signal on T2WI (Transverse position);C:High signal on FLAIR (Coronal position);D:No obvious enhancement in enhancement (Transverse position)
2.4基因检测病例1检测到LAMA2基因(NM_000426.3)1个杂合变异为50号外显子区域c.7147 C>T杂合突变,该突变为无义突变(p.Arg2383*),导致编码产物氨基酸序列提前终止,该突变来自于其父亲;另一突变为46号外显子区域c.6513_6515微缺失突变,引起所编码的蛋白质第2172位的氨基酸残基Val缺失,该突变来自于其母亲;此复合突变为已报道的致病突变。病例2检测到LAMA2基因(NM_000426.3)2个杂合变异,一个为46号外显子区域c.6513_6515微缺失突变,引起所编码的蛋白质第2172位的氨基酸残基Val缺失,该突变来自于其母亲;另一突变为17号内含子区域发生杂合突变c.2451-6A>G;p.?,该突变来自于其父亲,为国际上尚未报道的新发突变,未知意义。应用MLPA技术对LAMA2基因分别进行检测结果未见异常。病例3检测到LAMA2基因(NM_000426.3)2个杂合变异,一个为46号外显子区域c.7810C>T杂合突变,为无义突变(p.Arg2604*),导致编码产物氨基酸序列提前终止,该突变来自于其母亲,此突变为既往报道的致病突变;另一突变为24号内含子区域发生杂合突变c.3556-13T>A,该突变产生一个新的剪接受体位点,引起编码产物氨基酸序列提前终止,来自于其父亲。应用MLPA技术对LAMA2基因分别进行检测结果未见异常。
3 讨论
LAMA2-MD又称Merosin缺陷型先天性肌营养不良1A型(MDC1A),是一种常染色体隐性遗传性肌营养不良,是最常见的先天性肌营养不良(CMD),全球患病率(1~9)/10万人,欧美国家多见,在先天性肌营养不良中约占1/3[2,4-8]。LAMA2-MD发病的主要机制是肌肉层黏连蛋白α2的完全或部分缺乏。当层黏连蛋白α2有缺陷或缺失时,肌肉纤维受到机械应力,容易撕裂和断裂,导致组织损伤和变性[9]。LAMA2-MD其特征是出生时或出生后6个月内发病,表现为严重的肌无力和肌张力低下,以近段肌力低下为主,面肌受累出现吮吸无力,部分出现关节挛缩[10-15]。大多数患儿的运动发育里程碑延迟,2~3岁时会独坐,很少有人获得独立的行走[16]。LAMA2-MD的临床表现取决于层黏连蛋白α2缺乏的程度。层黏连蛋白-α2的完全缺失表现为严重的早发性CMD,而层黏连蛋白-α2的部分缺失则症状较轻,获得独走能力[17-19]。本组病例2肌肉病理示Merosin部分肌纤维膜表达减弱,局灶缺失,其起病相对较晚,症状也较轻,最终能独立的行走。病例3肌肉病理示Merosin表达明显减弱或缺失,出生后即出现严重症状,吮吸无力,发育里程碑落后,仅能独坐。本组3例患儿营养状态不良,体质量低下。喂养困难、吞咽异常和咀嚼困难是影响LAMA2-MD患儿体质量增加不良的原因。此外,反复感染进一步加剧了这一问题[20]。大多数早发性LAMA2-MD患儿体质量低于第三百分位,有些患儿需要肠内喂养以满足其营养需求[21-22]。
LAMA2-MD患儿精神状况正常,可有轻微智能发育迟滞或癫痫,言语认知功能通常正常,仅少数智力发育落后或合并癫痫,少数大脑皮质发育不良患儿伴难治性癫痫[23-24]。本组患儿智能发育均正常,脑电图检查均示正常儿童脑电图。层黏连蛋白-α2广泛分布于横纹肌的肌膜,同时也表达于脑血管的基膜及施万细胞基底膜,这种表达方式正是LAMA2-MD脑及周围神经系统病理基础[21]。6个月大的患者中可以观察到头颅典型脑白质病变,1岁后稳定无变化,这种表现对诊断很有帮助。本组3例患儿头颅MRI表现与既往报道一样,主要累及脑室旁和皮层下白质,而胼胝体和内囊不受累,呈长T1、长T2信号,FLAIR上呈高信号。这种改变可能缺陷影响血脑屏障的成熟和(或)功能的改变,脑血管基膜对水通透性增加,大脑含水量的变化有关[25]。此外,层黏连蛋白-α2在施万细胞基底膜均有表达,有报道其缺陷能影响周围神经髓鞘形成引起运动感觉神经病变,但临床意义不大[26]。
LAMA2-MD的致病基因LAMA2定位于6q22-23,包含65个外显子,编码分子量为390 000的laminin-α2[3]。LAMA2基因分子遗传学检测是确诊LAMA2-MD的最有效的方法,检测手段包括NGS联合MLPA、aCGH等[27]。LAMA2基因突变类型复杂多样,主要包括无义突变(32%)、微小缺失或插入(19%)、剪切突变(15%)、错义突变(12%)、拷贝数变异(20%)等[28-29]。两个LAMA2基因的功能缺失突变能导致更严重的早发型LAMA2-MD,这些突变分布在LAMA2编码区,55%聚集在14、25、26和27号外显子[17]。另一方面,错义突变、框内缺失和剪接位点突变常与晚发型LAMA2-MD相关,在这些病例仍然可以检测到层黏连蛋白-α2的残留表达。GERANMAYEH等[17]报道病例中2名患儿LAMA2基因拷贝虽然功能完全缺失,其临床表型却较温和,能够独立行走,不需要喂养或通气支持。本研究中病例1是既往已经报道过的临床病例[30]。病例2、3虽然根据肌肉活检组织病理检查所见已做出临床病理诊断,但结合免疫组织化学染色和LAMA2基因突变分析对明确诊断十分必要。2名患儿通过基因检查都检出2个LAMA2基因突变,分别来自于父亲和母亲的致病基因,符合常染色体隐性遗传遗传方式。病例2检出的一个突变是6号外显子区域c.6513_6515微缺失突变,为朱艳慧等[31]于2014年报道的致病突变,引起所编码的蛋白质第2172位的氨基酸残基Val缺失,影响laminin-α2与钙离子的结合,从而导致laminin-α2与α-抗萎缩相关糖蛋白连接受阻。另一突变为17号内含子区域发生杂合突变c.2451-6A>G,此突变为国际上尚无报道的新突变,结合免疫组化结果分析虽为未知突变,但该位点相对较为保守,推测这种突变能产生了一个新的剪接受体位点,导致编码提早终止,引起蛋白表达缺失。病例3检出的一个突变是46号外显子区域c.7810C>T杂合突变,为无义突变,引起编码产物氨基酸序列提前终止。2014年EGL基因诊断、Eurofins临床诊断已将该突变列为致病突变[27,32]。另一突变为24号内含子区域发生杂合突变c.3556-13T>A,既往43名层黏连蛋白α2缺陷型先天性肌营养不良报道中有2名患而同样有这种变异,认为该突变为致病突变,产生一个新的剪接受体位点,引起编码产物氨基酸序列提前终止[29]。
LAMA2-MD致残率和致死率高,预后差,目前缺乏有效根治性治疗方法,主要强调对患儿的多学科综合管理和长期随访[33-34]。基因检测可确定致病基因和遗传方式,对确诊的先证者家庭提供精准遗传咨询并进行产前诊断,对优生优育有重要指导意义。
