肝硬化、高血糖合并脑桥中央髓鞘溶解症1例报告
2020-08-07伍文清张静林黄宇明
寇 程,伍文清,张静林,张 磊,黄宇明
脑桥中央髓鞘溶解症(central pontine myelinolysis,CPM)是一种累及脑桥的渗透性髓鞘溶解,CPM病例最早见于1959年Adams报道,随着医学影像学的发展,使得该病得以更多的在临床中被报道[1]。现将我院收治的1例在发病1 m及46 d分别进行过头颅核磁共振(magnetic resonance imaging,MRI)的病例报道如下,分析CPM头颅MRI的动态演变。
1 临床资料
患者,男,46岁,因“左侧肢体无力、行走不稳1 m,加重2 d”于2018年2月20日收入院。既往乙肝病史40 y未治疗,长期酗酒,未戒酒。患者自2018年1月中旬出现左侧肢体力弱感,不能持重物,伴行走不稳。2018年2月出现左手指麻木、进食呛咳。入院前1 w觉吐字不清,入院前4 d开始斋戒,入院前2 d患者出现左手持水杯掉落、浑身疲倦、下地行走困难。入院前2 d曾有烦躁、大喊大叫、答非所问情况,持续约4 h好转,好转后遗留反应慢。入院当天头颅MRI示脑桥斑片样异常信号(见图1)、双侧底节区对称T1WI高信号(见图2)。
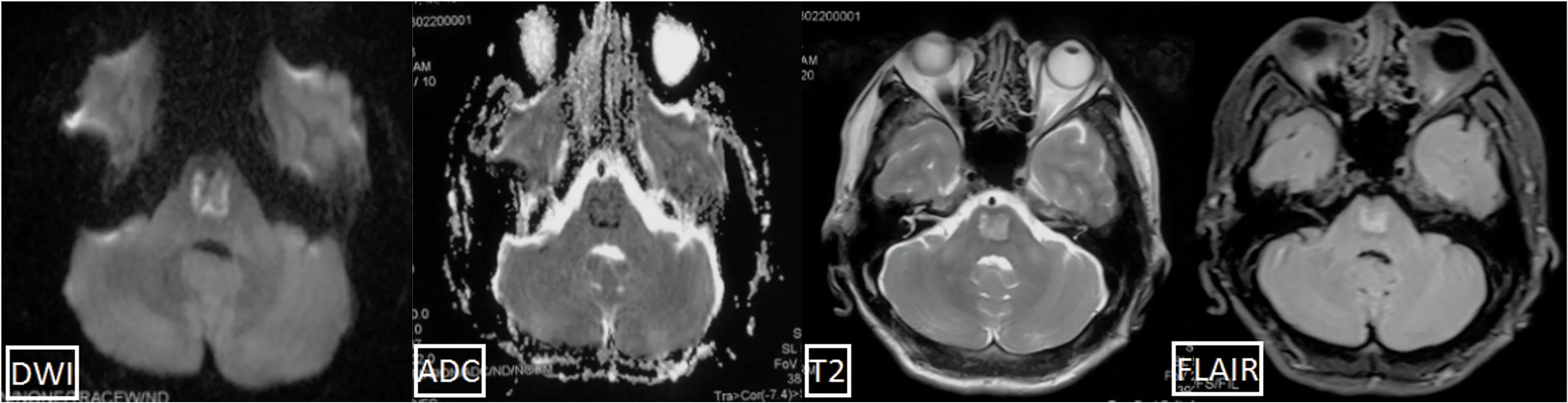
图1 发病1 m时头颅MRI:DWI、ADC可见病灶边缘水分子弥散受限。T2WI、FLAIR加权,脑桥可见斑片样高信号改变
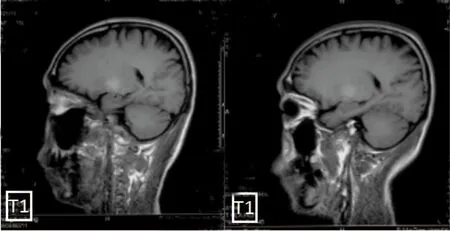
图2 发病1 m头颅MRI:双侧底节区对称T1WI高信号改变
入院查体:肝病面容,肝掌、蜘蛛痣阳性,全身皮肤黏膜、双侧巩膜黄染。腹软,未及压痛、反跳痛,脾大过中线,腹围80 cm。双下肢水肿。神清,构音障碍,颅神经查体阴性。右侧肢体肌力5级,左侧肢体肌力5-级,右手指鼻不稳,左手指鼻稍不稳,双下肢跟膝胫试验睁眼、闭眼均不稳,Romberg征睁眼、闭眼均不稳。感觉检查未见异常。四肢腱反射对称引出,双侧Babinski征阴性。入院当天化验血WBC 2.76×109/L(正常值 4×109/L~10×109/L),RBC 3.39×1012/L(正常值4×1012/L~5.5×1012/L),HGB 117 g/L(正常值120~160 g/L),PLT 57 ×109/L(正常值100×109/L~300×109/L),血钠 132.4 mmol/L(正常值137~147 mmol/L),血氯97.4 mmol/L(正常值99~110 mmol/L),血钾4.77 mmol/L(正常值3.5~5.3 mmol/L),血糖27.87 mmol/L(正常值3.6~6.1 mmol/L),血氨 27 μmol/L(正常值10~47 μmol/L),血浆晶体渗透压304.5 mOsm/(kg·H2O)[正常值298.7 mOsm/(kg·H2O)],丙氨酸氨基转移酶ALT 441.7 U/L(正常值9~50 U/L),门冬氨酸氨基转移酶AST 478 U/L(正常值15~40 U/L)。给予胰岛素泵入降糖,同时给予复方甘草酸苷保肝治疗、静脉补钾、奥美拉唑保护胃黏膜治疗,当天共计静脉摄入氯化钠8 g。入院第二天化验回报如下:WBC 1.66×109/L,RBC 2.64×1012/L,HGB 91 g/L,PLT 21.2×109/L。凝血酶原时间PT 13.8 s(正常值9.4~12.5 s),凝血酶原活动度PTA 73%(正常值70%~130%),国际标准化比值(INR) 1.28(正常值0.8~1.2)。ALT 323.3 U/L,AST 380.6 U/L,总胆红素 53.7 μmol/L(正常值0~18.8 μmol/L),直接胆红素 28.9 μmol/L(正常值0~6.8 μmol/L),白蛋白21.4 g/L(正常值40~55 g/L),胆碱酯酶1732 U/L(正常值4000~11000 U/L)。血钾 3.26 mmol/L,血钠 141.1 mmol/L,血氯107.1 mmol/L,血尿素 4.49 mmol/L(正常值3.1~8 mmol/L),血肌酐 44.2 μmol/L(正常值57~97 μmol/L),血糖 10.67 mmol/L,血浆晶体渗透压300.99 mOsm/(kg·H2O)。尿糖+,尿酮体+。糖化血红蛋白12.6%(正常值4.6%~6.2%)。甲胎蛋白2 ng/ml (正常值0.89~8.78 ng/ml)。HBV DNA 9.13×106IU/ml(正常值<20 IU/ml)。腹部超声:肝硬化,脾大,腹水,门脉高压,胆囊壁增厚,脾-肾分流,双侧胸水。脑电图示正常范围脑电图。MMSE(大专)28分。入院后根据患者亚急性病程,主要为进行性加重的左侧肢体无力、双下肢行走不稳,伴有进食呛咳、言语不清,存在酗酒、肝硬化、禁食的诱因,神经系统查体主要为小脑及脑干受损的体征,结合患者头颅核磁存在脑桥中央斑片样T2WI、FLAIR高信号、T1WI低信号,DWI、ADC序列病灶病缘存在水分子弥散受限,患者影像表现符合脑桥中央髓鞘溶解症改变,故考虑患者为CPM。给予患者恩替卡韦抗乙肝病毒,多烯磷脂酰胆碱、复方甘草酸苷、还原型谷胱甘肽保肝治疗。给予胰岛素强化降糖。给予叶酸、B1、甲钴胺补充维生素营养神经治疗。静点白蛋白补充白蛋白,呋塞米、螺内酯利尿治疗腹水。治疗20 d后患者症状好转出院。出院时情况:神清语利,颅神经查体阴性。左下肢轻瘫试验阳性,余肢体肌力5级,双手指鼻稍不稳、左下肢跟膝胫试验不稳,闭目难立征阴性。四肢腱反射,双侧巴氏征阴性。复查头颅MRI脑桥ADC低信号消失(见图3),底节区T1WI高信号同前(见图4)。
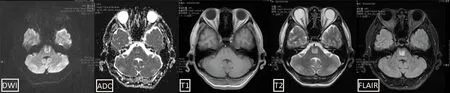
图3 发病46 d头颅MRI:脑桥ADC低信号消失,DWI、T2WI、FLAIR仍可见高信号改变,T1WI为低信号改变
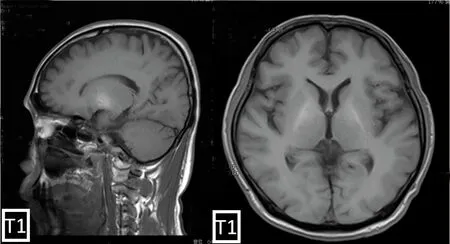
图4 发病46 d头颅MRI:双侧底节区对称性T1WI高信号改变较图2无变化
2 讨 论
CPM病例最早见于1959年Adams报道,CPM的组织病理学特点是髓鞘内分裂、空泡化和髓鞘破裂,而轴索保留,呈非炎症性脱髓鞘[1]。CPM在MRI上表现为脑桥基底部长T1长T2信号,无明显强化表现、无占位效应、病灶多为对称分布[2]。本例患者脑桥病变对称分布、无占位效应,信号改变与CPM相符,故诊断为CPM。常规头颅MRI序列(T1WI、T2WI、FLAIR)在髓鞘溶解2 w后才能出现异常信号,CPM的潜在病因是水电解质失衡导致的渗透紊乱,DWI序列对水的运动较敏感,DWI序列在CPM临床症状出现的24 h内就可出现高信号改变[2]。在DWI出现高信号区域对应ADC出现低信号,证实存在水分子弥散受限[1,2],在临床症状出现1 m左右,这种弥散受限信号逐渐减弱[1],本例患者在发病1 m时头颅MRI仍可见到病灶边缘有少许弥散受限表现,发病46 d左右病变区域ADC低信号消失,DWI、T2WI、FLAIR信号较前无变化,这提示ADC序列对CPM病程变化更敏感。
既往有关低钠血症相关的CPM报道较多,本例患者无严重低钠血症及快速纠正低钠血症病史。文献中对于无电解质紊乱而由于长期高血糖、高血糖高渗状态以及纠正高血糖过程中发生CPM的病例也有报道[3],本例患者入院后随机血糖27.87 mmol/L,糖化血红蛋白12.6%且该患者入院前并不知晓自己患糖尿病、未监测过血糖及降糖治疗。因此,考虑此例患者由于高血糖导致CPM可能性较大。关于高血糖所致CPM的原因文献中报道可能与高血糖导致的高渗透压进而破坏血脑屏障,尤其是在长期不控制血糖的糖尿病患者中,高的渗透压会导致大脑少突胶质细胞的蛋白损伤和聚集[3]。
本例患者头颅MRI除脑桥异常信号外,双侧底节区苍白球附近也存在对称性T1高信号改变,因其信号改变与脑桥外髓鞘溶解(extrapontine myelinolysis,EPM)MRI表现的T1等信号或低信号不同,故除外EPM。EPM底节区病灶急性期在MRI表现为双侧尾状核头及壳核DWI、T2WI、FLAIR高信号,ADC低信号,T1WI可无改变;病程1 m时可表现为T1WI低信号,T2WI、FLAIR、DWI高信号,ADC低信号消失[1]。既往研究[4,5]显示70%~100% 肝硬化患者可出现双侧苍白球对称性T1WI高信号改变,此种异常可能与肝硬化门脉分流导致肝脏对于血清锰的清除力下降,进而导致锰在苍白球的蓄积有关。
CPM迄今尚无特异性治疗,其病程一般较长且潜在死亡风险高,CPM患者大多合并有严重的基础疾病,比如肝衰竭、肾衰竭、恶性肿瘤等,治疗多为对症支持治疗及基础疾病治疗,因此对于该病的预防更重要[1]。
综 述
