经基因诊断的周围神经病样肯尼迪病1例报告
2020-08-07方招弟李志刚徐武华张君臣钟水生
方招弟,李志刚,徐武华,张君臣,钟水生
肯尼迪病(Kennedy disease,KD)是一种临床罕见的X连锁隐性遗传性神经元变性疾病。在既往的文献中,因神经损伤主要累及脊髓和延髓下运动神经元,故又被称为脊髓延髓肌萎缩症(spinal and bulbar muscular atrophy,SBMA)[1]。近年来,国内外陆续发现以非下运动神经元受损为首发或主要临床症状的个案[2~4],但鲜有周围神经病样型报道。今提供1例我科近期确诊的该型个案汇报如下。
1 临床资料
患者,男,53岁,广州市郊区农民。因进行性四肢乏力10 y,伴言语含糊、呛咳、行走不稳5 y于2019年12月4日入院。患者于10 y前首先出现进行性加重的双下肢乏力,突出表现为长距离步行和下蹲起身吃力,但否认有间歇性跛行、大小便控制困难以及步行不稳等症状。随后出现双手持重物费力,无法胜任日常田间劳作和基本家务,同时伴随四肢、躯干和面颊等部位肌肉的逐渐萎缩、不断加频的无痛性肌跳以及无肿胀感和触痛的双乳增大。近5 y渐出现行走不稳,需小心翼翼,并经常看路面,同时伴有吐词不清、饮水呛咳、咀嚼无力、食道梗阻感和醉酒样头晕等症状。
患者已婚,育2子,体健,否认家族中有类似症状患者。体格检查:血压136/80 mmHg,神志清楚,对答切题,构音不清,高级神经功能检查正常。双侧眼裂、额纹和鼻唇沟对称。双侧咽反射完全消失,伸舌居中,舌缘略呈齿状并可见明显的舌肌纤颤。肩胛、躯干及四肢和面部,尤其是大小鱼际肌、掌指骨间肌可见明显的肌萎缩(见图1A~D),肌固有反射减弱,肌张力大致正常,近端肌力IV级,远端肌力V-级。四肢腱反射减弱,上下肢锥体束征阴性。双手意向性震颤,步态不稳,走直线不能,Romberg征:睁眼(-)闭眼(+),加强征(+)。痛觉、温度觉、震动觉、位置觉均减退。明显的乳房发育(见图1E),但体毛分布及外生殖器未见异常。

图1 患者肌萎缩、乳房发育情况(A~E)
辅助检查:三大常规、肝肾功能、空腹和餐后血糖、血脂、甲状腺功能、各种炎症、肿瘤和传染病指标以及心电图、全腹部彩超和胸片均未见明显异常。肌酸激酶升高(637.7U/L,正常参考值50~310U/L),但肌酸激酶同工酶正常。性激素五项:促黄体生成素9.17 IU/L(参考值1.7~8.6 IU/L),垂体催乳素14.69 μg/L(参考值4.04~15.2 μg/L),促卵泡生成素7.29 IU/L(参考值1.5~12.4 IU/L),雌二醇117.50 pmol/L(参考值94.8~223 pmol/L),睾酮7.82 μg/L(参考值1.93~7.40 μg/L)。头颅MR和MRA无明显异常发现,颈腰椎MR提示颈 3/4、颈 4/5、腰 3/4、腰 4/5 椎间盘轻度膨出。
神经电生理检查结果:肌电图:(1)运动神经传导:左上肢尺神经,右侧正中神经 DL延长,CMAP降低;双下肢腓总神经、胫神经MCV减慢,DL延长,CMAP降低;2)感觉神经传导:双侧正中神经、左侧尺神经SCV减慢,SNAP 降低;右尺神经SNAP未测出,左侧腓浅神经SNAP降低,腓肠神经SCV减慢,SNAP 降低;右侧腓浅神经,腓肠神经均未测出;(3) 神经电图:左下肢胫神经、左侧正中神经F波、H反射潜伏时延长,右下肢胫神经、右侧正中神经F波未测出;(4)针极肌电图:右侧斜方肌、左侧胸锁乳突肌、双侧拇短展肌、左侧骨间肌、左侧肱二头肌、双下肢胫前肌、腓肠肌有少量纤颤、正尖电位,MUP 增宽,募集降低。
基因检测:雄激素受体(AR)基因第1外显子中的三核苷酸(CAG)重复序数为50次,属于全突变范围(见图2)。
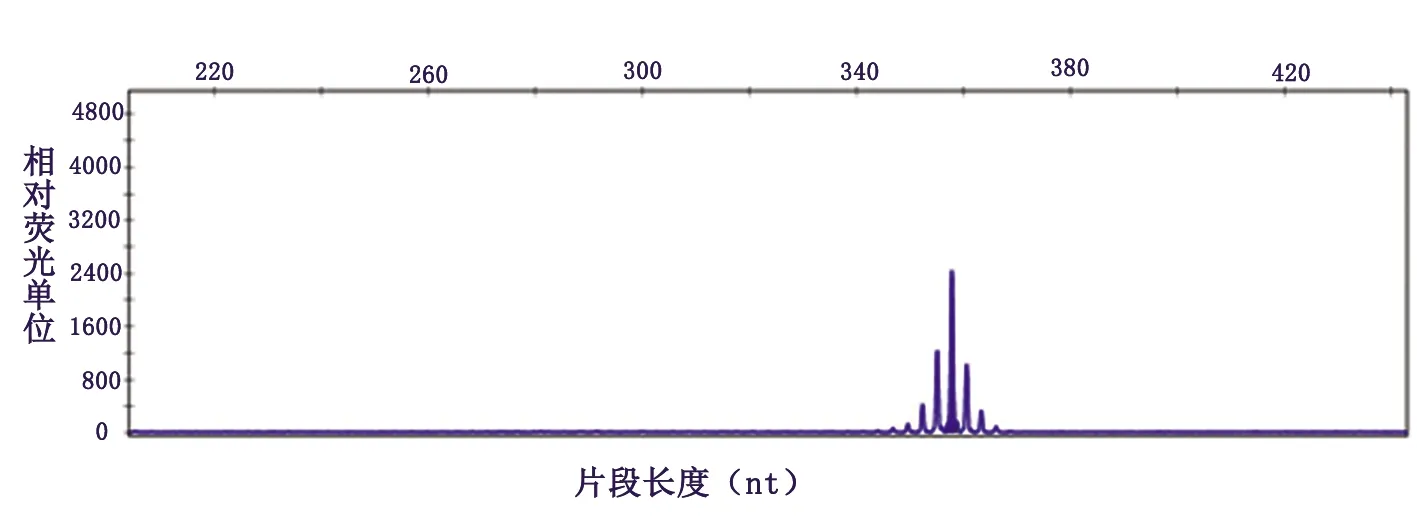
图2 患者血样本雄激素受体基因第 1 外显子中 CAG 重复序列数为50
治疗:患者因个人原因未接受抗雄激素治疗,仅接受一周的神经营养类药物及对症治疗,症状未见明显变化。
2 讨 论
本例患者的临床存在以下特征:(1)缓慢起病,进行性加重;(2)症征主要累及肌肉运动和感觉系统,且肌萎缩明显;(3)肌电图检查提示广泛的运动和感觉神经受损;(4)明显的男性乳房发育。结合其高达50次的AR基因第1外显子CAG重复次数,符合2011年修订的KD确诊标准[5]。
本例患者的肌无力发展模式与国内外文献报道的绝大多数KD患者一样,自下往上,逐步发展至舌面部,就诊时虽四肢肌肉萎缩明显,仍能保持一定的机动性[6]。值得注意的是,近年来陆续出现缺乏明显肌无力和肌萎缩征的非经典个案,如以手颤[7]、延髓肌无力[8]、肌痛[9]、肢体感觉异常[10]、乳房发育[11]或性功能障碍[8]等为首发症状者。值得一提的是,本例患者临床上肌萎缩、肌束震颤远较无力症状更为明显,且伴随着肌固有反射、腱反射、深浅感觉的减弱,而其肌电图检查并未发现经典的下运动神经元受损征象,强烈提示其神经损伤部位为周围神经。
KD的发病机制迄今未明。在以下几种可能的机制:(1) 配体依赖的AR毒性作用;(2)线粒体功能损伤所致氧化应激;(3)自噬性改变;(4)AR激活功能区域-2调节功能受损等[12]中更多指向第一种机制:转基因KD模型雄性小鼠能成功复制出KD样临床症状,并在体内检出大量的突变AR蛋白,而接受药物抗雄激素治疗或者阉割后,其临床症状可被纠正[13],这与临床上广泛观察到的男性患者居多以及AR突变基因女性携带者多无明显症状的现象相符。鉴于AR广泛分布于全身各个脏器,结合近期文献报道的多样性病理损伤,本病例进一步支持这一观点:KD很可能不是一种传统意义上的单纯下运动神经元变性疾病,更可能是一种多系统受累的全身性疾病[14,15]。目前尚不清楚CAG重复序列数目与临床之间的关联。较为肯定的是,CAG重复序列数目与发病年龄呈负相关,即重复次数越多,发病年龄越早。而重复次数与临床症状和病情严重程度之间的关系尚存争议,国外学者更倾向于正相关联[15],而国内学者则多持无关论[16,17]。目前我们尚不清楚该患者症状的未来走向,但从其现有的临床表象来看,不排除进一步发展出下运动神经元受损的可能,有待于后期的随访加以证实。
目前KD尚无有效的治疗方法,重点在于并发症的预防,抗雄激素药物治疗亮丙瑞林在2期临床试验中已取得部分成效[13],而基因治疗[18]及肌肉导向疗法[6]被视为潜在有效的治疗选项。
