7,8-二甲氧基-2,3,4,5-四氢-1H-苯并[c]氮杂卓盐酸盐的简便合成
2020-07-22张永霞郑银平靳清贤张洪培
张永霞,郑银平,靳清贤*,张洪培
(1.河南交通职业技术学院 公共基础教学部,河南 郑州 450005;2.郑州轻工业大学 材料与化学工程学院,河南 郑州 450001;3.郑州衍生生物科技有限公司,河南 郑州 450001)
苯并氮杂卓盐酸盐类化合物具有非常多样的药理活性,如有较强的抗病毒、抗惊厥活性、抗癫痫活性等[1],尤其在治疗神经系统疾病领域,特别是与多巴胺D1和D3受体相关的药物成瘾领域中的应用[2],许多药物分子和天然产物中都含有苯并氮杂卓盐酸盐结构或片段,因而苯并氮杂卓盐酸盐衍生物是重要的药物中间体[3]。目前文献报道较为普遍的制备方法多以酮类为起始物,经贝克曼重排得到酰胺,酰胺还原后制得苯并氮杂卓(Scheme 1)[4-7],但此类方法用到易爆物,有一定安全隐患。Alonso等报道了一种以3,4-二甲氧基苯甲醛为起始原料,与苯硫醚丙胺通过希夫碱、还原、保护、氧化、成环、脱硫等多步反应制备苯并氮杂卓[8],其步骤繁琐,且涉及含硫物质,有较大气味。Horiguchi等报道的以邻二氯苄与脂肪胺类成环法,收率低,仅有22%[9]。

Scheme 1
有文献报道直接以3,4-二甲氧基苯丙胺为起始原料,经过草酰氯制备成异氰酸酯,然后在多聚磷酸催化下成环,再经硼烷还原得到游离的7,8-二甲氧基-2,3,4,5-四氢-1H-苯并[c]氮杂卓(Scheme 2)[10]。但中间体异氰酸酯不易制备(收率66%),且利用多聚磷酸催化异氰酸酯成环时副产物多,收率只有40%,效果不佳。
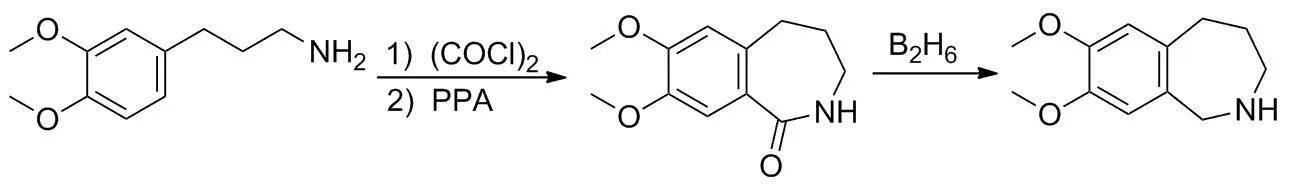
Scheme 2
本文以3,4-二甲氧基苯丙胺为主,进行路线设计,该路线如下[11]:以3,4-二甲氧基苯丙酸为起始原料,经酯化、氨解、还原、酰化保护、成环、脱酰基、Boc保护、脱保护成盐得到目标产物分子,共八步反应,合成路线如Scheme 3所示。所用原料及试剂均为廉价易得物质,所用的合成方法简便,各步骤收率高,产物易纯化。

Scheme 3
1 实验部分
1.1 仪器与试剂
RY-2 型显微熔点仪;Avance 400 MHz型超导核磁共振仪(DMSO-d6或CDCl3为溶剂,TMS为内标);Thermo Finnigan LCQ Advantage MAX型质谱仪。
所用试剂均为分析纯。
1.2 合成
(1) 3,4-二甲氧基苯丙酸甲酯(1)的合成
称取3,4-二甲氧基苯丙酸4.21 g(0.02 mol)溶于60 mL无水甲醇中,滴加氯化亚砜2.39 g(0.02 mol),滴毕,升温至70 ℃,反应6 h,旋转蒸发除去多余溶剂,残余倒入冰水中,二氯甲烷萃取3次,合并有机层,依次用碳酸氢钠溶液洗涤,无水硫酸钠干燥,过滤后旋蒸除去溶剂。得油状液体4.16 g,收率93%;1H NMR(400 MHz,CDCl3)δ:2.60(t,2H),2.90(t,2H),3.66(s,3H),4.86(s,6H),6.76(m,3H);HR-MS(ESI)m/z:Calcd for C11H14O4{[M+H]+}224.25,found 225.26。
(2) 3,4-二甲氧基苯丙酰胺(2)的合成
称取13.36 g(0.015 mol)溶解于氨水/甲醇=1/1(30 mL)中,升温至80 ℃,反应10 h(TLC监测)。冷冻反应液,有结晶析出,过滤干燥得白色固体22.92 g,收率93%[12],m.p.122~123 ℃;1H NMR (400 MHz,CDCl3)δ:2.52(t,2H),2.93(t,2H),3.86(s,3H),3.87(s,3H),5.40(s,2H),6.72~6.82(m,3H);HR-MS(ESI)m/z:Calcd for C11H15NO3{[M+H]+}209.24,found 210.26。
(3) 3,4-二甲氧基苯丙胺(3)的合成
称取22.09 g(0.01 mol)溶于40 mL四氢呋喃中,再加入四氢锂铝0.76 g(0.02 mol),升温至80 ℃,反应10 h(TLC监测)。冷却至室温,旋蒸除去THF,缓慢加水后加硫酸镁,抽滤,滤饼用乙酸乙酯洗涤,滤液用乙酸乙酯萃取3次,合并有机层,旋蒸浓缩得淡黄色液体31.56 g,收率80%[13];1H NMR(300 MHz,CDCl3)δ:6.68~6.78(m,3H),3.83(s,3H,),3.82(s,3H),2.70(t,2H),2.58(t,2H),1.70~1.75(m,2H),1.17(br s,2H,NH2);HR-MS(ESI)m/z:Calcd for C11H17NO2{[M+H]+}195.26,found 196.26。
(4)N-乙酰基-3,4-二甲氧基苯丙胺(4)的合成
称取31.56 g(0.008 mol)溶于40 mL二氯甲烷中,滴加三乙胺1.65 g(0.016 mol),反应置于冰水浴,然后滴加乙酰氯0.79 g(0.0096 mol),25 ℃反应2 h(TLC监测)。加水洗涤分液,有机层减压除溶得淡黄色液体42.39 g,收率96%[14];1H NMR(400 MHz,CDCl3)δ:6.68~6.78(m,3H),3.83(s,3H),3.82(s,3H),2.70(t,J=7.0 Hz,2H),2.58(t,J=7.2 Hz,2H),1.84(s,3H),1.70~1.75(m,2H);HR-MS(ESI)m/z:Calcd for C13H19NO3{[M+H]+}237.29,found 238.29。
(5)N-乙酰基-7,8-二甲氧基-2,3,4,5-四氢-1H-苯并[c]氮杂卓(5)的合成
称取41.75 g(0.007 mol)溶于30 mL的硫酸中,多聚甲醛0.42 g(0.014 mol),加热至60 ℃反应5 h(TLC监测)。反应体系倒入冰水中,乙酸乙酯萃取3次,乙酸乙酯层用碳酸钠溶液洗涤一次,将乙酸乙酯旋蒸浓缩,淡黄色液体51.35 g,收率78%,化合物未纯化直接用于下一步。
(6) 7,8-二甲氧基-2,3,4,5-四氢-1H-苯并[c]氮杂卓(6)的合成
称取51.49 g(0.006 mol)溶于40 mL甲醇中,加入碳酸钾1.66 g(0.012 mol),升温至70 ℃反应12 h(TLC监测)。旋蒸除去溶剂,残余加适量水和乙酸乙酯萃取,得油状液体61.23 g,收率84%;1H NMR(400 MHz,CDCl3)δ:6.65(s,1H),6.34(s,1H),3.82(s,6H),3.02~2.99(m,2H),2.72~2.68(m,2H),1.80~1.76(m,2H),1.62~1.59(m,2H);HR-MS(ESI)m/z:Calcd for C12H17NO2{[M+H]+}207.27,found 208.26。
(7)N-Boc-7,8-二甲氧基-2,3,4,5-四氢-1H-苯并[c]氮杂卓(7)的合成
称取61.26 g(0.006 mol)溶于40 mL的二氯甲烷中,加入Boc酸酐1.44 g(0.0066 mol)和三乙胺0.909g(0.009 mol),室温反应1 h,然后加水,水层用二氯甲烷萃取两次,合并二氯甲烷层,残余经柱层析(洗脱剂:乙酸乙酯/石油醚=10/1,V/V)纯化得油状液体71.82 g,收率93%;1H NMR(400 MHz,DMSO-d6)δ:9.13(s,2H),7.07(s,1H),6.88(s,1H),4.21(s,2H),3.75(s,3H),3.73(s,3H),3.31~3.24(m,2H),2.89(d,2H),1.82(s,2H),1.38(s,9H);HR-MS(ESI)m/z:Calcd for C17H25NO4{[M+H]+}307.38,found 308.39。
(8) 7,8-二甲氧基-2,3,4,5-四氢-1H-苯并[c]氮杂卓盐酸盐(8)的合成
称取71.63 g(0.005 mol)溶于二氯甲烷,通入氯化氢气体,反应2 h,体系逐渐析出固体,过滤得到白色鳞片状固体81.11 g,收率92%;1H NMR(400 MHz,DMSO-d6)δ:9.13(s,2H),7.07(s,1H),6.88(s,1H),4.21(s,2H),3.75(s,3H),3.73(s,3H),3.31~3.24(m,2H),2.89(d,2H),1.82(s,2H);HR-MS(ESI)m/z:Calcd for C12H18NO2Cl{[M+H]+}243.73,found 208.39;Anal.calcd for C12H18NO2Cl:C 59.14,H 7.39,N 5.75,Cl 14.58,found C 59.26,H 7.37,N 5.69,Cl 14.58。
2 结果与讨论
2.1 1的合成
本步属于经典氯化亚砜催化的酯化反应,反应后处理简便且收率高。此方法优于硫酸酯化法,因为硫酸酯化后处理会产生较多废酸污水。本步反应加热能更快的促进反应转化,相比室温,可大大缩短反应时间。
2.2 2的合成
本步采用氨水直接进行胺解法制备酰胺,研究发现室温下反应进行极其缓慢,需要数天,因此将反应加热可缩短反应时间。但是温度不宜过高,过高或导致酯水解形成羧酸。经研究对比发现反应温度80 ℃为宜,反应10 h氨解基本完全,同时产物析出,后处理简便。
2.3 3的合成
本步采用经典的四氢锂铝还原酰胺成伯胺,唯一缺点是反应处理比较麻烦,因为四氢锂铝产生的絮状物导致萃取难以分层,过滤堵塞滤纸,经借鉴硫酸镁处理法,可以让反应后处理体系结成沙粒状后,易于抽滤。
2.4 4的合成
酸催化成环要求伯胺需要进行乙酰化保护,否则成环无法发生,乙酰化我们采用乙酰氯,并用三乙胺做缚酸剂,反应可在室温下顺利发生。
2.5 5的合成
本步借鉴四氢异喹啉六元环成环方法,以硫酸为催化剂,多聚甲醛为碳源,成功延伸至七元环的成环合成,方法成熟可靠,收率中等。
2.6 6的合成
成环后的N-乙酰基-7,8-二甲氧基-2,3,4,5-四氢-1H-苯并[c]氮杂卓需要将乙酰基脱去,本文采用碳酸钾在甲醇里加热脱除,反应时间略长。采用强碱氢氧化钾可以缩短反应时间,但是由于氢氧化钾碱性过强,且反应需要加热,或导致产物收率降低,综合考虑,我们采用弱碱碳酸钾,延长时间,增加收率。
2.7 7的合成
由于得到的游离态7,8-二甲氧基-2,3,4,5-四氢-1H-苯并[c]氮杂卓为液体,薄层层析极性较大,不易纯化,且游离的胺类物质不易存放,而做成盐酸盐易于保存,引入Boc保护能实现此目的,因此对游离态7,8-二甲氧基-2,3,4,5-四氢-1H-苯并[c]氮杂卓进行Boc保护,保护以后薄层层析极性减小,易于纯化,纯化后可方便制得纯净的盐酸盐。
2.8 8的合成
Boc保护的7,8-二甲氧基-2,3,4,5-四氢-1H-苯并[c]氮杂卓在经过柱层析纯化后,经过氯化氢气体脱除Boc,同时游离胺与盐酸结合形成盐酸盐,为白色鳞片状晶体,易于干燥存放。
3 结论
提供了一种7,8-二甲氧基-2,3,4,5-四氢-1H-苯并[c]氮杂卓盐酸盐简便制备方法,并对分步反应过程进行了分析讨论,筛选优化反应条件,该路线适合此类物质的批量制备。
致谢:抑菌和除草活性测试由南开大学元素有机化学国家重点实验室生物活性测试室完成,谨表谢意!
