一种基于风车格结构的有效降低内重组能的咔唑类格子化分子
2020-07-13史海涵吴香萍彭辛哲余国静董朝阳纪瑶瑶杨思文陈俊林冉雪芹解令海
史海涵, 吴香萍, 彭辛哲, 余国静, 董朝阳, 纪瑶瑶, 杨思文,陈俊林, 王 锦, 冉雪芹, 杨 磊, 解令海, 黄 维,,3
(1. 南京邮电大学有机电子与信息显示国家重点实验室培育基地, 江苏省生物传感材料与技术重点实验室,材料科学与工程学院, 海外教育学院, 南京 210023;2. 南京工业大学柔性电子重点实验室, 先进材料研究院, 南京 211816;3. 西北工业大学柔性电子研究院, 西安 710072)
环番是一种具有芳烃单元的大分子, 含有特定的功能单元、多个反应位点以及一定尺寸的空腔, 因而具有良好的分子识别能力、光学性质、催化活性及药物活性等[1~5]. 但由于环番缺少刚性正交的骨架, 因此很难实现延伸位点的平行分布. 而芴是一种具有刚性的分子, 因此, 将环番中的“环”与芴结合, 设计了“格子化”结构, 得到了日字格[6]、口字格[7~9]、井字格[10,11]和风车格[12,13]等多反应位点的刚性格分子. 其中, 风车格具有2组相互平行且对称的刚性分子骨架, 合成较为简单. 虽然这种风车格分子具有较好的深蓝光发射性能, 但是由于芴的9号位易被氧化而出现绿光带[14], 导致发光不太稳定.
含氮杂环因为其独特的发光优势引起了关注, 其中咔唑作为一类富电子的含氮杂环化合物, 具有独特的较大的π-共轭刚性平面结构, 属于类芴体系, 在光电材料、染料及超分子识别等领域有着潜在的应用价值[15~20]. 咔唑基团具有良好的给电子能力, 且结构易被修饰, 被引入其它分子结构中能够提高化合物的稳定性和玻璃转化温度(Tg), 可用于设计高稳定性、高效率的蓝光和绿光材料[15,21]. 除此之外, 咔唑本身及其衍生物通常均具有良好的空穴传输特性[22,23], 且其电子与空穴能够处于良好的平衡状态[16].
因此, 本文将4个咔唑分子通过2,9号位相互连接设计成为一个风车格, 并研究了该咔唑类格子化分子的结构特点及热力学和电子性质.
1 理论计算方法
所有量子化学计算均采用Gaussian 09程序[24]进行. 采用密度泛函理论(DFT)的B3LYP杂化泛函结合6-31G(d)基组对研究体系的基态、阳离子及阴离子态几何结构进行优化. 通过计算获得了化合物的电离势(IP)、电子亲和势(EA)、最高占据分子轨道(HOMO)、最低未占据分子轨道(LUMO)和能隙(Eg)等.
为了研究分子内的非共价相互作用, 对其进行了约化密度梯度(RDG)分析. RDG是Yang等[25]提出的一种弱相互作用的可视化分析方法, 为小分子、分子复合物和固体中的空间相互作用提供了有效表征. 利用MULTIWFN软件[26]绘制了两个函数之间的散点图, 然后用VMD软件[27]绘制彩色RDG等值面图. RDG(r)(a.u.)定义如下:
(1)
式中:ρ(r)(a.u.)为电子密度;ρ(r)为电子密度梯度. 此外, RDG填色等值面图可以清楚地指示分子中空间相互作用的位置, 通过对不同颜色的区分可以看出各种非共价相互作用的类型. 为了进行更深入研究, 还对二聚咔唑(DZP)、三聚咔唑(TZP1~3)以及风车格分子组成的超分子(Dimer 1和Dimer 2)进行了RDG分析, 并进行了对比.
重组能(λ)在电荷转移过程中起着至关重要的作用, 可以分为内重组能和外重组能, 后者主要受周围介质的影响, 其贡献非常微弱, 常常被忽略. 重组能可以由四点法和正则振动分析(NM)两种方法计算得到. 如果忽略介质的极性和分子振动因素的影响, 四点法计算所得重组能只与垂直跃迁能量差有关(图1).
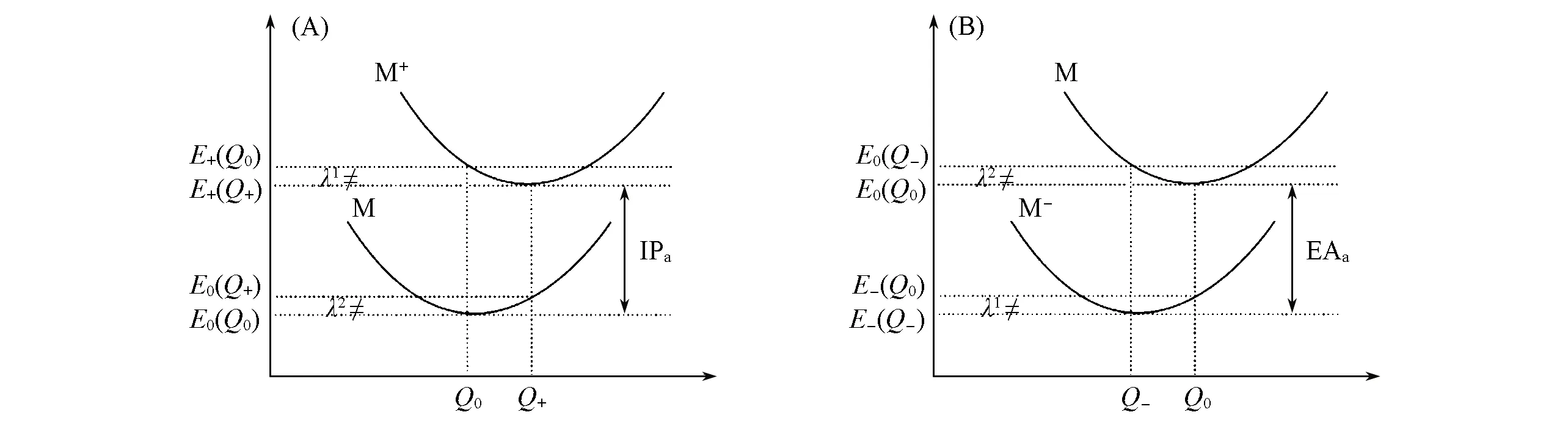
Fig.1 Schematic diagram of four-point method for hole(A) and electron(B) transfer
由图1可见,E0(Q0)表示基态分子的中性状态,E+(Q0)表示阳离子分子的中性状态,E+(Q+)表示阳离子分子的阳离子状态,E0(Q+)表示基态分子的阳离子状态,E-(Q0)表示阴离子分子的中性状态,E-(Q-)表示阴离子分子的阴离子状态,E0(Q-)表示基态分子的阴离子状态, 空穴重组能λ(h)(eV)可通过下式计算:
λ(h)=λ1+λ2=[E+(Q0)-E+(Q+)]+[E0(Q+)-E0(Q0)]
(2)
电子重组能λ(e)(eV) 通过下式计算:
λ(e)=λ1+λ2=[E-(Q0)-E-(Q-)]+[E0(Q-)-E0(Q0)]
(3)
另外, 绝热电离势(IPa, eV)和绝热电子亲和势(EAa, eV)的计算如下:
IPa=E+(Q+)-E0(Q0)
(4)
EAa=E-(Q-)-E0(Q0)
(5)
在基于电荷状态和中性状态极小点结构的正则模式来做分解的NM分析方法中, 重组能被分解为不同振动模式对分子重组能的贡献. 重组能由Dushin程序分析拆解[28]. 在谐波近似下, 其重组能(λ, eV)被描述为
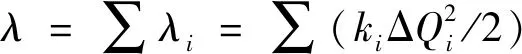
(6)
式中:λi(eV)对应频率为ωi的模式i的重组能;ki为相应振动模式下的力常数; ΔQi为沿分子电荷状态和中性状态的平衡位置间的正常模式坐标的位移. 通过对每个振动模式相对应的各个重组能求和来获得总重组能. NM分析方法能够深入理解分子不同的振动模式对分子重组能贡献的差异; 然而, 谐振模型只是对实际势能面的近似, 所以两种方法计算的重组能并不能精确相等.
2 结果与讨论
2.1 分子结构和前线分子轨道分析
图2为风车格分子(GZP)的结构示意图, 密度泛函理论(DFT)计算结果显示, GZP含有2种构象(图3), 与环己烷的两种极限构象(船式与椅式构象)类似. 先从两种构象的能量角度来分析, 咔唑风车格的船式构象GZP1(0 kJ/mol)能量比椅式GZP2(122.88 kJ/mol)低, 所以GZP1较稳定. 其中, GZP1构象的内孔径为0.298 nm, 外孔径为1.079 nm, 而GZP2构象的内外孔径分别为0.290和1.209 nm.
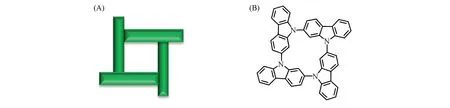
Fig.2 Schematic diagram of the organic molecules with shape of the windmill(A) and the molecular structure(B) of GZP
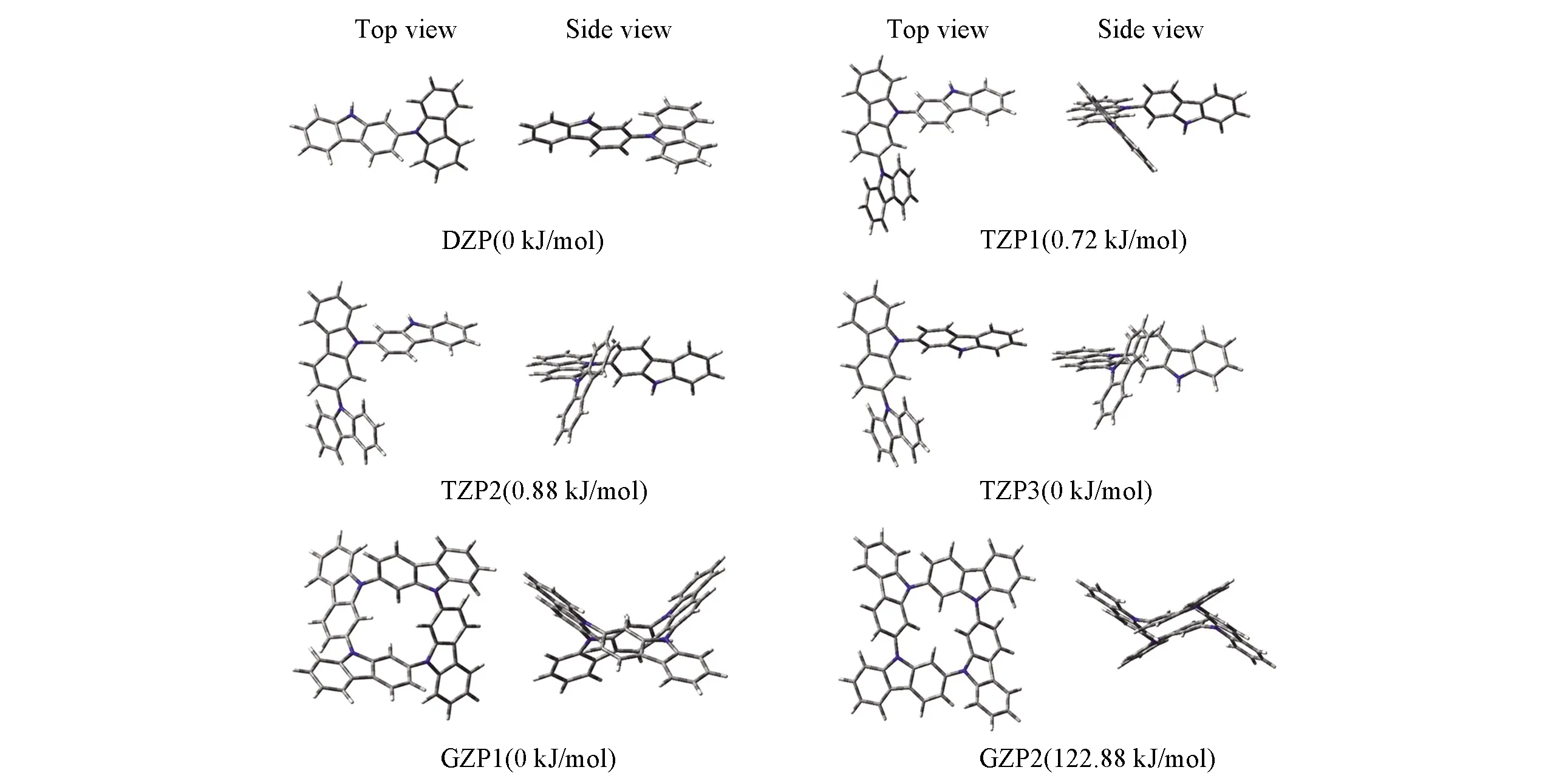
Fig.3 Top and side view of the optimized geometries of DZP, TZPs and GZPs with the corresponding energy levels
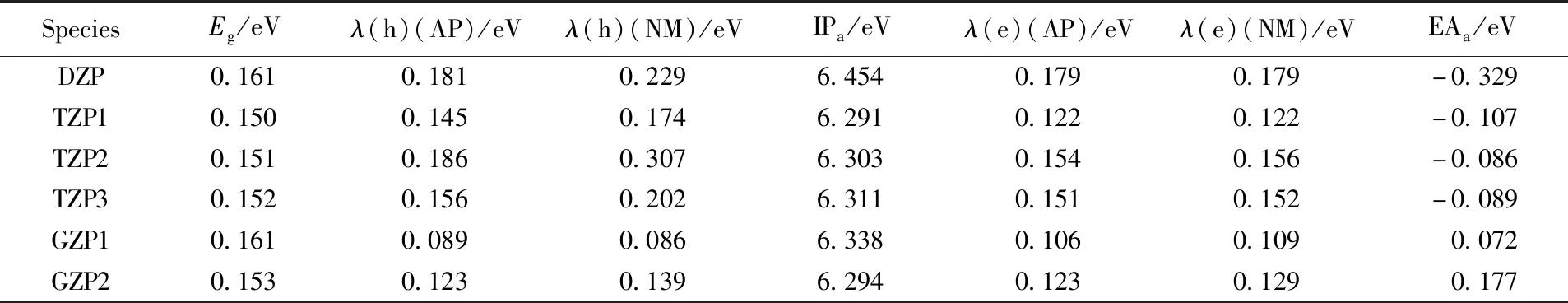
前线分子轨道可以用来分析有机化合物的电子和光学性质[29]. HOMO和LUMO在化学反应中起着极其重要的作用. HOMO轨道能量对应于IP, 而LUMO轨道能量对应EA. 图4为二聚咔唑、三聚咔唑及GZP1和GZP2的HOMO, LUMO图. 可见, DZP的HOMO分布在整个分子骨架上, 且分布不太均匀, LUMO分布在其中一个咔唑分子上. TZP的HOMO分布在由2个咔唑单元形成的长链一侧, 而LUMO则分布在另一个咔唑单元上. 咔唑单元形成格分子后, GZP1的HOMO, LUMO轨道和GZP2的HOMO轨道均匀分布在整个分子骨架上, 而GZP2的LUMO轨道分布不均匀, 其电子云主要分布在对角线上的2个咔唑单元, 这些轨道分布不仅可以解释分子的HOMO-LUMO能隙, 还可以解释分子化合物的稳定性[30]. 表1列出了分子的HOMO-LUMO能隙(Eg). 可见, GZP1和GZP2的能隙分别为0.161和0.153 eV, 说明GZP1的化学活性较低, 更加稳定.
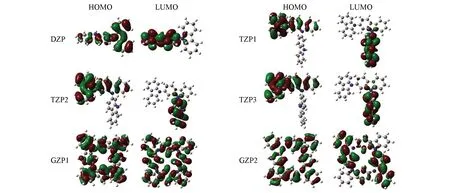
Fig.4 Frontier molecular orbitals(HOMO and LUMO) of DZP, TZPs, GZPs
2.2 可视化非共价相互作用分析
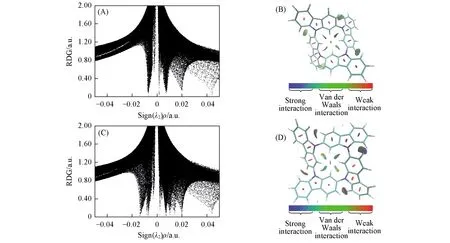
Fig.5 Scatter plots of the reduced density gradient(RDG)(A,C) and the RDG isosurface(B,D) for GZP1(A,B) and GZP2(C,D)
讨论了咔唑单元组成的DZP, TZP1~3, GZP1和GZP2两种构象异构体及其超分子Dimer1和Dimer2的非共价相互作用. 图5为GZP1和GZP2的RDG散点图以及相应的RDG等值面图, RDG散点图中的峰值表示弱相互作用, 图中横坐标Sign(λ)ρ的正负分别表示排斥和吸引相互作用[31]; 其中, 红色区域表示存在强烈排斥作用, 蓝色区域表示存在强吸引相互作用, 绿色区域表示存在弱范德华相互作用. RDG等值面图直观地显示了分子中空间相互作用的位置和强弱. 在DZP的RDG散点图(图S1, 见本文支持信息)中有4个峰值, 分别为±0.007, 0.02和0.044 a.u., 其中, 0.02和0.044 a.u.附近的峰表示苯环和五元环中心区域的空间排斥相互作用, 这些空间排斥相互作用在RDG等值面上用红色的椭球形表示; ±0.007 a.u.的尖峰对应RDG等值面上绿色和棕色混合区域, 表明了两个相邻咔唑单元之间的弱相互作用力; 对比DZP, TZP1~3的峰值没有任何变化, 相应的RDG图中作用力的性质没有变化, 只是增加了数量. 但形成格子后, GZP1和GZP2均在0.002 a.u.附近额外增加了一个尖峰, 对应RDG等值面上分子中心的绿色区域, 对应咔唑单元在形成格子后中心区域出现的弱相互作用. 而GZP2在0.01和±0.014 a.u.附近额外增加了3处尖峰, 这是由于GZP2的咔唑单元之间的排列出现差异, 咔唑单元之间夹角不同, 从而导致弱相互作用强度不同. 而Dimer1和Dimer2均在±0.002 a.u.出现了多个尖峰, 这对应于2个分子间出现的弱相互作用.
2.3 电离势、电子亲和势和重组能分析
对于电荷传输材料, 分子的IP和EA是评估空穴和电子传输的重要参数, 在传输材料中, 空穴传输层(HTL)的IP越低, 铟锡氧化物(ITO)的空穴越易进入空穴传输层; 电子输运层(ETL)的EA越高, 电子越易从阴极进入到ETL[32]. 表1列出了分子的IPa和EAa的计算结果, 对比DZP和TZP, GZP的IPa值没有太大的变化, 而EAa值明显增大, IPa值的大小顺序为TZP1
重组能是决定有机半导体电子转移速率的一个重要参数. 根据Marcus理论, 较低的重组能对应较高的电荷迁移率. 重组能分为内重组能和外重组能, 前者衡量电子得失后或电子态改变后因几何结构的弛豫而导致的体系能量的变化, 后者对应的是将周围环境分子重新极化所消耗的能量. 在晶体或固体中, 由于环境相对固定外重组能可以被忽略[33]. 用绝热势能面(AP)法和NM法计算了分子内重组能. DZP, TZP1~3, GZP1和GZP2的λ(h)和λ(e)计算结果列于表1,λ(h)的大小顺序为GZP1 Fig.6 Contributions of vibrational modes to the relaxation energy[λ(1)] for DZP(A), TZP1(B), TZP2(C), TZP3(D), GZP1(E) and GZP2(F) Table 1 Internal reorganization energies calculated by AP method and NM analysis of DZP, 设计了一个由4个咔唑组成的GZP格分子, 其具有刚性的分子结构和弱的分子内相互作用, 有利于其化学稳定性. 通过AP法和NM法分析评估的GZP的内重组能一致. GZP1的内重组能非常低, 空穴和电子传输过程中的内重组能分别为0.089 和0.106 eV. 研究结果表明, 格子化结构能有效降低了重组能, 可以有效改善有机半导体材料的电荷转移速率. 支持信息见http://www.cjcu.jlu.edu.cn/CN/10.7503/cjcu20200054.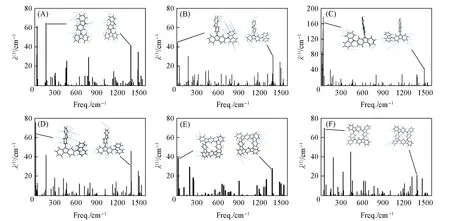

3 结 论
