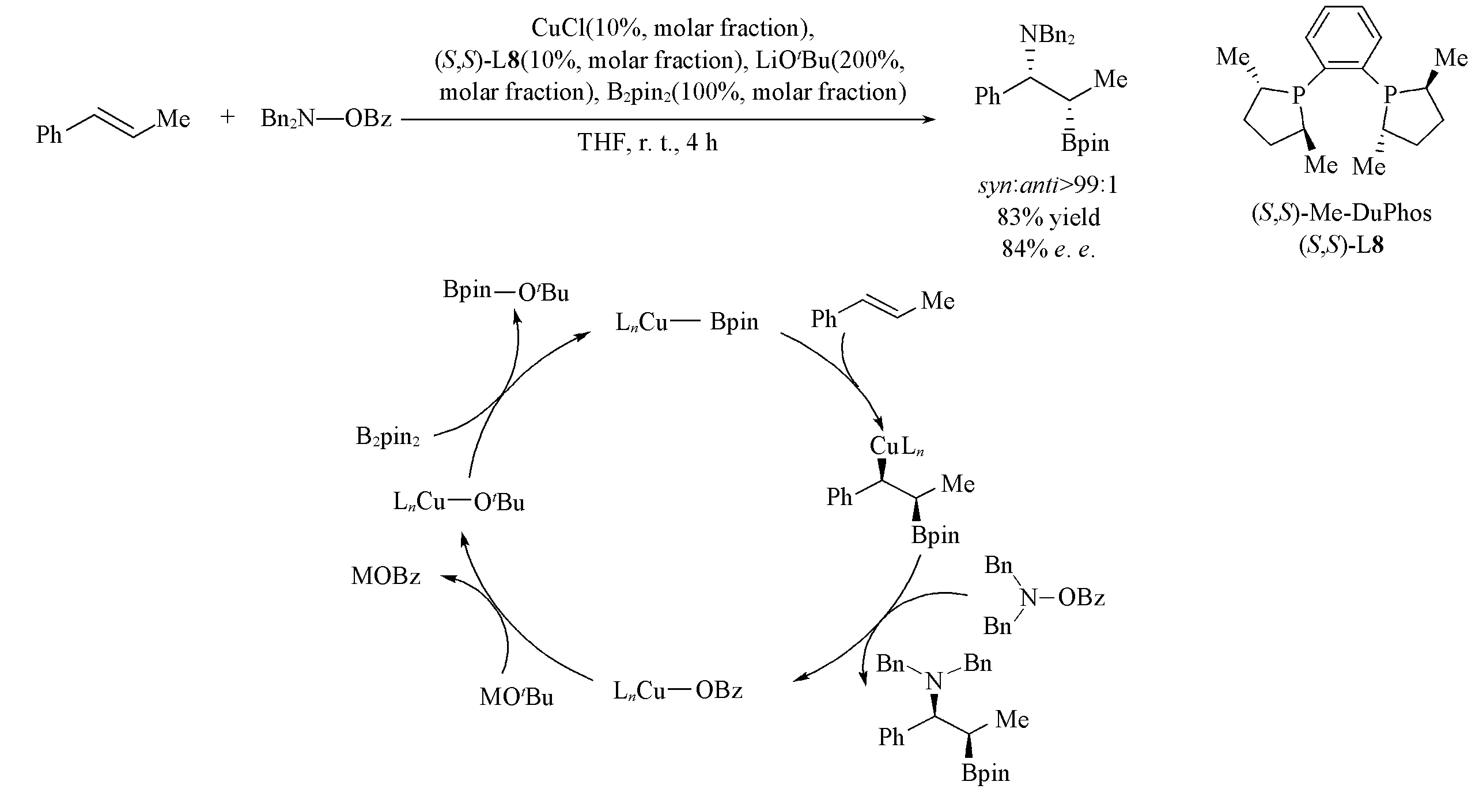
催化不对称碳硼成键反应研究进展
2020-07-13黄明耀朱守非
黄明耀, 朱守非
(南开大学化学学院, 元素有机化学研究所, 天津 300071)

Scheme 1 Applications of chiral organoboron compounds(A) Transformations of boryl groups; (B) chiral organoboron pharmaceuticals.

Scheme 2 Asymmetric catalytic constructions of C—B bond
有机硼化合物在有机合成[1,2]、生物医药[3,4]及材料科学[5,6]领域中均有重要应用. 其中, 手性有机硼化合物不仅能在转化为芳基、烯基、羟基、羧酸及氨基等官能团时保持构型[Scheme 1(A)], 而且自身也能作为药物或生物活性分子用于多种疾病的诊断和治疗[Scheme 1(B)], 使得手性有机硼化合物的高效精准合成成为有机化学的重点研究方向. 本文从不对称催化的角度综合评述了C—B键的构筑方法, 即使用手性催化剂实现的对映选择性碳硼成键反应, 不包括手性原料的衍生化、手性拆分及手性辅基参与的合成等使用化学计量手性试剂的碳硼成键反应.
按照反应类型, 催化条件下C—B键的不对称构筑方法主要可分为硼试剂对不饱和键的不对称加成反应、不对称C—B键偶联反应、金属卡宾对B—H键的不对称插入反应及其它不对称碳硼成键反应等(Scheme 2).
1 硼试剂对双键的不对称加成反应
1.1 不对称硼氢化反应
烯烃硼氢化反应指B—H键对烯烃双键的加成反应. 如果烯烃两端的取代基不同, 则反应存在区域选择性和立体选择性问题, 使用催化剂可以实现选择性调控. 自1988年Burgess等[7]报道了Rh(Ⅰ)搭配双磷配体催化的烯烃不对称硼氢化反应以来, 各类过渡金属催化的烯烃不对称硼氢化反应得到了广泛研究. 目前, 已有较多文献对这一方向的研究进展做了比较系统的综合评述[8~10].
1.2 不对称硼化质子化反应
2008年, Yun等[11]发现CuCl与二茂铁骨架手性双磷配体L1的络合物作为催化剂, 对α,β-不饱和腈类或酯类化合物的硼化质子化反应有良好的不对称诱导效果, 可以得到单一的β-硼化产物[Scheme 3(A)]. Marder和Lin等[12]通过计算深入研究了该反应的机理, 他们认为Cu催化剂先与联硼烷发生复分解生成活性的Cu-B物种, 由于该物种中硼更具负电性, 因而在后续烯烃迁移步骤中硼加成到β位形成α-Cu羰基中间体, 这也是该反应的手性决定步骤. 该中间体可能通过2种途径完成催化循环: (1) 直接与体系中的质子源反应生成产物, 同时重生Cu前体; (2) Cu迁移到氧上, 然后与联硼烷发生复分解反应重生Cu-B催化剂, 释放出的烯醇硼酯经后处理水解后得到硼氢化产物. 与传统的硼氢化反应反应不同, 该反应中的硼与氢分别来自于联硼烷与外加质子源, 因而可称为硼化质子化反应[Scheme 3(B)].

Scheme 3 Cu(Ⅰ)-catalyzed asymmetric protoboration of alkenes(A) and proposed mechanism(B)
双磷配体[13~17][L1, Scheme 4(A); L2, Scheme 4(B)]、双胺配体[18][L3, Scheme 4(C)]、氮杂环卡宾(NHC)配体[19,20][L4, Scheme 4(D)]及联吡啶配体[21][L5, Scheme 4(E)]等多种手性配体相继被用于Cu催化缺电子烯烃的不对称硼化质子化反应. 除了α,β-不饱和脂或α,β-不饱和腈, 该反应还适用于α,β-不饱和醛酮及α,β-不饱和酰胺. 当使用β,β-二取代的缺电子烯烃时, 可构筑季碳手性中心.
Scheme 4 Cu-catalyzed asymmetric protoboration of alkeneswith different ligands (A) Bisphosphorous ligand L2; (B) bisphosphorous ligand L1; (C) diamine ligands; (D) NHC ligands; (E) bipyridine ligands.
当缺电子烯烃β位连有烯基时, 有可能发生1,6-加成生成δ-硼化的产物. 2013年, Kobayashi等[22]报道了Cu(Ⅱ)与联吡啶类配体L5络合物催化的β-烯基取代环烯酮的不对称硼化质子化反应, 该反应在水相中进行, 能生成单一的δ-硼化产物, 对映体过量值(e.e.)可达89%[Scheme 5(A)]. 值得注意的是, 这类水相反应体系与Cu(Ⅰ)催化的有机相反应体系不同, Cu以水溶性更好的Cu(Ⅱ)离子形式参与催化循环而不是被还原为Cu(Ⅰ)[23]. 2014年, Lam等[24]采用Cu(Ⅰ)与双磷配体L1络合物作为催化剂实现了链状共轭二烯1,6-硼化质子化, 高对映选择性合成了δ-硼化产物[Scheme 5(B)].

Scheme 5 Cu(Ⅱ)/L5-catalyzed asymmetric 1,6-protoboration of α,β,γ,δ-unsaturated compounds(A) and Cu(Ⅰ)/L1-catalyzed asymmetric 1,6-protoboration of α,β,γ,δ-unsaturated compounds(B)
上述体系都是通过手性配体修饰的金属络合物作为催化剂来调控反应的对映选择性, 当底物中有活性较高的醛基时, 可以通过手性胺催化剂活化缺电子烯烃, 进而实现反应的手性诱导. 2011年, Ibrahem和Córdova等[25]通过天然脯氨酸衍生的手性胺C1对底物醛基的活化作用, 结合Cu(Ⅱ)/PPh3催化体系, 实现了对α,β-不饱和醛的高对映选择性β-硼化反应. 该催化过程经历一个亚胺中间体, Cu—B对烯烃加成时的面选择性由胺上的手性环境来调控. 他们还将所得硼化产物通过一步Wittig反应转化为手性δ-硼取代酯(Scheme 6).

Scheme 6 Cu/amine-cocatalyzed asymmetric protoboration of α,β-unsaturated aldehyde
除了缺电子烯烃, Cu-B物种还可用于催化其它类型烯烃的硼化质子化反应. 2009年, Hoveyda等[26]报道了NHC配体L6或L7与Cu(Ⅰ)络合物催化的苯乙烯衍生物的不对称硼化质子化反应, 得到了在芳基β-位硼化的产物[Scheme 7(A)]. 2010年, Ito等[27]报道了Cu/双膦配体L8络合物催化的1,3-二烯的不对称硼化质子化反应. 上述反应在不同温度下给出不同的化学选择性: 在温度较低时反应倾向于先发生1,2-加成生成烷基-Cu中间体, 而后与醇通过六元环协同过程质子解得到形式上的1,4-加成产物, 表现出动力学选择性; 而温度较高时, 1,2-加成后的烷基-Cu中间体则易于发生1,3-Cu迁移而形成更稳定的1,4-加成中间体, 随后与质子解生成形式上的1,2-加成产物, 表现出热力学选择性[Scheme 7(B)]. Tortosa等[28,29]报道了Cu-B催化的环丙烯与环丁烯的不对称硼化质子化反应.

Scheme 7 Cu(Ⅰ)-catalyzed asymmetric protoboration of aryl ethylenes(A) and Cu(Ⅰ)-catalyzed asymmetric protoboration of conjugated dienes(B)
Cu-B催化体系还能用于一些不饱和氮杂环类化合物的硼化质子化反应. Ito等[30]报道了Cu(Ⅰ)/双磷配体L9催化的缺电子吲哚类化合物的不对称硼化质子化反应, 得到吲哚3位顺式硼化的产物,e.e. 值可达98%[Scheme 8(A)]. 2016年, 他们[31]通过两步一锅法实现了吡啶类化合物的不对称还原/硼化反应: 第一步使用硼氢化钠将吡啶去芳构化, 部分还原至二氢吡啶; 第二步是反应的关键步骤, 在Cu(Ⅰ)/双膦配体L2的催化下, 能够在二氢吡啶的3,4位发生反式硼化质子化反应, 得到高e.e. 值的反式产物[Scheme 8(B)]. 类似地, 他们也实现了喹啉类化合物的还原/不对称硼化质子化反应[32].

Scheme 8 Cu(Ⅰ)-catalyzed asymmetric protoboration of indoles(A) and Cu(Ⅰ)-catalyzed asymmetric protoboration of pyridines(B)
不仅是烯烃, Cu-B催化的硼化质子化过程同样适用于碳-杂原子双键. 由于Cu-B物种中硼基团具有一定亲核性, 因而它更易于进攻碳杂原子双键中相对缺电子的碳而形成碳端硼化的产物, 从而实现C—B键的构筑. 2013年, Tian和Lin等[33]报道了Cu/手性NHC配体L10催化的亚胺硼化质子化反应, 该反应对芳香醛和脂肪醛衍生的亚胺都表现出中等或较好的对映选择性[Scheme 9(A)]; 该反应的机理与烯烃硼化质子化过程类似, 决定化学选择性与对映选择性的步骤是碳氧双键对Cu—B键的迁移插入. 2015年, Liao等[34]引入了双齿的磷-亚砜类配体, 其对芳基亚胺的硼化质子化反应具有良好的不对称诱导效果; 在其它条件相同的情况下, 使用不同的Cu(Ⅰ)前体能够得到绝对构型相反的产物, 他们认为这是由于共价型与离子型Cu催化剂生成的Cu-B物种构型上存在差异引起的[Scheme 9(B)]. 2015年, Ito等[35]报道了一例醛羰基的硼化质子化反应: 使用手性双膦配体L12, 能够高对映选择性合成α-羟基硼, 该产物与氯硅烷反应可转化为更稳定的α-硼基硅醚[Scheme 9(C)].

Scheme 9 Cu(Ⅰ)/L10-catalyzed asymmetric protoboration of imines(A), Cu(Ⅰ)/L11-catalyzed asymmetric protoboration of imines(B) and Cu/L12-catalyzed asymmetric protoboration of aldehydes(C)
一些强Lewis碱, 如NHC等, 能与联硼酸酯上的一个硼络合从而使另外一个硼基团表现出一定的亲核性, 进而以硼负离子的形式加成到缺电子的烯烃的β位, 形成的中间体被质子化得到产物. 当使用手性的NHC催化剂时, 则能够在硼转移步骤控制其面选择性, 进而取得高对映选择性.
2012年, Hoveyda等[36]实现了手性NHC(C2)催化的缺电子烯烃的不对称硼化质子化, 该反应对α,β-不饱和酯、酰胺乃至环酮类底物都能够取得良好的收率及对映选择性[Scheme 10(A)]. 他们认为反应经历了如下历程: 首先Lewis碱NHC与联硼烷形成加合物, 活化了B—B键, 而后该加合物对缺电子烯烃进行1,4-加成, 亲核硼基团加到β位, 带NHC的亲电硼基团加到了氧端, 再经醇解得到产物, 同时再生NHC催化剂[Scheme 10(C)]. 2014年, Hoveyda等[37]将底物进一步拓展到位阻更大的β-取代的环酮类化合物, 得到手性的三级硼酸酯[Scheme 10(B)].

Scheme 10 C2-catalyzed asymmetric protoboration of alkenes(A), C3-catalyzed asymmetric protoboration of alkenes(B) and proposed mechanism(C)
1.3 不对称硼官能团化反应
与硼氢化的定义类似, 不饱和键的硼官能团化反应是指在双键两端分别加上硼基团与一个其它的官能团. 按照官能团的不同, 可以分为不对称双硼化反应、硼胺化反应及硼碳化反应.
1.3.1 不对称双硼化反应 由于双硼化合物中的2个硼都能够发生丰富的官能团转化, 而且有可能利用2个硼基团的性质差异(如电性、位阻等)实现选择性的转化过程, 因而其在有机合成中有着特殊的应用价值. 不饱和键的双硼化反应是合成双硼化合物的重要方法之一.
早期研究中, 烯烃的双硼化反应主要通过Pt, Pd及Rh等贵金属催化来实现, 一般认为其机理与硼氢化类似, 经过金属的氧化还原过程: 首先发生联硼试剂对金属中心的氧化加成形成金属双硼物种, 然后烯烃迁移插入到金属-硼键中, 最后经还原消除得到双硼化产物(Scheme 11), 反应的对映选择性取决于迁移插入步骤的面选择性控制. 反应中通常能够观察到部分反式加成产物, 其生成机理可能是金属碳中间体的β-氢消除后, 烯烃构型翻转后重新插入到金属-氢键中.
2003年, Morken等[38]报道了一例Rh(Ⅰ)催化烯烃的不对称双硼化反应. 采用P, N配体QUINAP(L13)/Rh(Ⅰ)催化体系, 1,2-二烷基取代烯烃能够与联儿茶酚硼烷发生顺式双硼化反应, 并能给出几乎光学纯的产物[Scheme 12(A)]. 反应同样适用于反式1,2-芳基烷基乙烯, 但是相对于顺式烯烃的反应效果差. Morken等[39]在后续的工作中对该反应进行了系统研究.

Scheme 12 Rh(Ⅰ)-catalyzed asymmetric diboration of 1,2-disubstituted alkenes(A), Pd(0)-catalyzed asymmetric diboration of allenes(B), Pt(0)-catalyzed 1,4-diboration of conjugated dienes(C) and Pt(0)-catalyzed 1,2-diboration of conjugated dienes(D)
2004年, Morken等[40]报道了Pd催化的联烯的不对称双硼化反应. 使用由Taddol衍生的手性亚磷酰胺配体L14与Pd的络合物作为催化剂, 单取代联烯的1,2-位能够发生不对称双硼化反应, 生成了同时具有烷基硼与烯基硼结构的手性双硼化合物[Scheme 12(B)]. 在后续研究中, Morken等[41]认为反应中Pd—B键更倾向于加成到联烯的2, 3位, Pd加到位阻较小的端位, 而后形成烯丙基Pd物种, 最后还原消除发生在1位形成1,2-加成产物.
2009年, Morken等[42]报道了一例Pt催化的烯烃不对称双硼化反应. 使用Taddol衍生的手性次磷酸脂配体L15与Pt的络合物作为催化剂, 共轭二烯能够与联频哪醇硼烷发生1,4-双硼化反应[Scheme 12(C)]. 反应经历一个烯丙基Pt中间体, 其还原消除时的区域选择性与烯丙基两端位阻有关, 当4-位位阻较大时硼化发生在2-位形成1,2-加成产物.
随后Morken等[43]通过底物的设计, 采用相似的催化体系实现了共轭二烯的不对称1,2-双硼化反应, 合成了一系列手性烯丙基硼化合物[Scheme 12(D)]. 除了共轭二烯, Morken等[44,45]也报道了对多种端烯和烯基硼的不对称双硼化反应, 取得了很好的手性控制效果. 此外, 他们[46]还实现了亚胺的不对称双硼化反应, 能够高效合成手性的α-氨基硼化合物.
除了经典的氧化还原机制, 一些研究者认为贵金属在催化烯烃的双硼化反应过程中可能不发生价态改变. 2013年, Nishiyama等[47]报道了Rh(Ⅲ)/手性双噁唑啉配体L17催化的烯烃的不对称双硼化反应, 对一系列端烯, 包括烷基端烯展现出良好的手性控制效果[Scheme 13(A)]. 他们认为, 在碱的作用下Rh(Ⅲ)与联硼酸酯反应生成单硼Rh(Ⅲ)催化剂, 之后烯烃迁移插入得到烷基Rh中间体, 该中间体可能直接与联硼酸酯发生转金属化完成催化循环, 也可能与体系中的单硼酸酯反应生成产物与叔丁氧基Rh(Ⅲ), 然后叔丁氧基Rh(Ⅲ)与联硼酸酯作用重生单硼Rh(Ⅲ)催化剂[Scheme 13(B)].
Scheme 13 Rh(Ⅲ)-catalyzed asymmetric diboration of alkenes(A) and proposed mechanism(B)

Scheme 14 Alcohol-catalyzed asymmetric diboration of alkenes (A) C4-catalyzed diboration; (B) C5-catalyzed diboration; (C) proposed mechanismof C4-catalyzed diboration; (D) proposed mechanismof C5-catalyzed diboration.
手性Lewis碱除了用于缺电子烯烃的硼化质子化反应之外, 还能够催化烯烃的不对称双硼化反应. 2012年, Gulyás和Fernández等[48]使用2倍摩尔量的手性苄醇C4作促进剂, 实现了端烯的不对称双硼化反应, 取得了最高42%(e.e.)的对映选择性[Scheme 14(A)]. 他们认为该反应经过协同过程: 手性苄醇作为Lewis碱活化了联硼烷, 然后通过四元环的过渡态一步形成了双硼化的产物[Scheme 14(C)]. 2016年, Morken等[49]通过使用手性二醇TBS-DHG(C5)与联硼烷B2(neo)2将该类反应的对映选择性进一步提高到了90%(e.e.)以上[Scheme 14(B)]. 但他们认为反应机理首先是手性二醇与联硼烷进行酯交换生成活性更高的B2(TBS-DHG)2(由于B—O键的动态平衡所致), 其在体系中游离烷氧基的活化下与端烯发生双硼化反应, 最后再次发生酯交换生成产物[Scheme 14(D)].
1.3.2 烯烃的不对称硼碳化反应 当烯烃迁移插入到金属硼物种中形成金属烷基中间体后, 可用各类碳基亲电试剂对其进行捕获, 从而生成烯烃的硼碳化产物. 与烯烃的硼氢化类似, 此类反应大多经历Cu-B中间体, 烯烃迁移插入过程是其区域选择性与对映选择性的决定步骤.
2008年, Ito和Sawamura等[50]报道了Cu(Ⅰ)/L2催化的不对称γ-硅基烯丙醇酯类化合物分子内硼碳化反应, 得到了手性的反式硅基-环丙基-硼类化合物(Scheme 15). 他们认为该反应首先经历烯烃对Cu-B物种迁移插入过程, 由于Si基团上的Si—Me键对邻位Cu—C键的超共轭稳定作用(α-硅效应), Cu物种被导向地加在氧的γ位, 再通过构象调整发生分子内亲核取代反应环化形成产物[Scheme 15(B)]. 他们进一步将定位基团从硅基换为苯基[51], 也能够经过类似的环化过程, 高对映选择性地得到反式产物.

Scheme 15 Cu(Ⅰ)-catalyzed asymmetric intramolecular carboration of alkenes(A) and proposed mechanism(B)
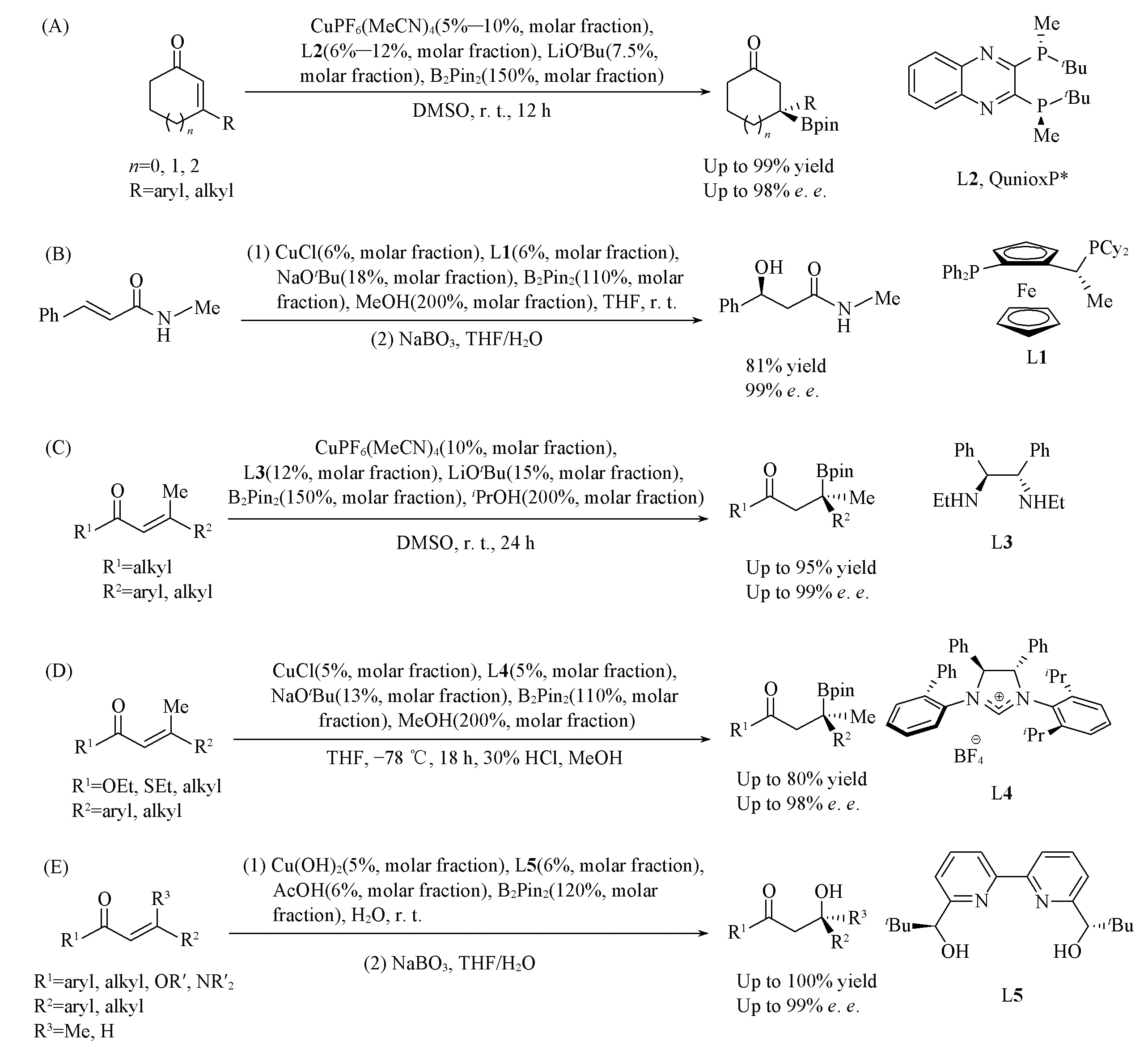
Scheme 16 Cu(Ⅰ)-catalyzed asymmetric allylic boration of alkenes
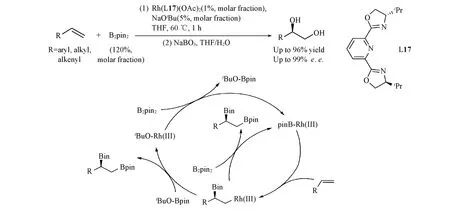
除了分子内反应, Cu-B体系还可用于催化分子间的烯烃不对称硼碳化反应. 2014年, Hoveyda等[52]报道了Cu/L18催化的联烯不对称硼烯丙基化反应(Scheme 16). 通过手性Cu-B物种对联烯的选择性加成, Cu加在端位形成烷基-Cu中间体, 该中间体对高活性的烯丙醇磷酸酯经由八元环过渡态发生γ-取代反应, 生成相应的γ-手性烯基硼化合物. 该反应被成功用于天然产物Rottnestol和Herboxidiene的克级规模不对称全合成中.
2015年, Liao等[53]利用Cu/Pd双金属催化体系, 实现了苯乙烯类化合物的不对称硼烯丙基化反应[Scheme 17(A)]. 该反应以接力的形式进行: 手性亚砜与Cu(Ⅰ)催化体系可高效控制苯乙烯类底物的迁移插入过程, 进而高对映选择性地形成Cu-烷基中间体, 该中间体以保持构型的形式对烯丙基Pd物种转金属化, 再经还原消除即可得到β-手性有机硼化合物[Scheme 17(B)]. 利用类似的双金属催化体系, Brown等[54]实现了芳基取代内烯的不对称硼芳基化反应. 2018年, Liao等[55]采用Cu/亚砜和Cu/双膦配体催化体系, 实现了甲基碘作亲电试剂的烯烃不对称硼甲基化反应.

Scheme 17 Cu/Pd co-catalyzed asymmetric allylic borylation of alkenes(A) and proposed mechanism(B)
Scheme 18 Cu-catalyzed asymmetric carboration of allenes with aldehydes and ketones(A) and Cu-catalyzed asymmetric carboration of allenes with imines(B)
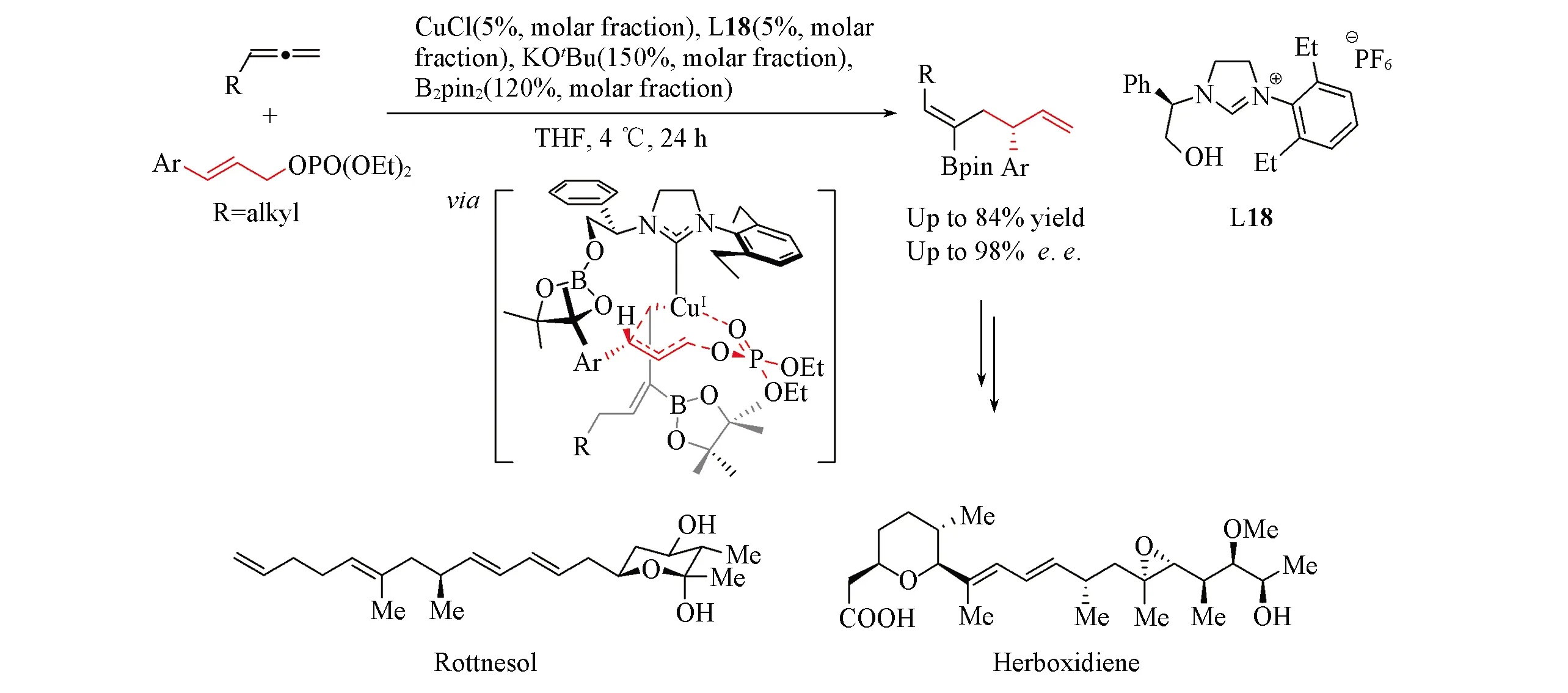
对于一些极性的不饱和键, 如醛、酮、亚胺或Michael受体等, 同样可以作为亲电试剂来高效地捕获烷基-Cu物种. 2013年, Hoveyda等[56]报道了Cu/双磷配体L20或L21催化的联烯的不对称硼-羟烷基化反应[Scheme 18(A)]. 该反应同样是由联烯对Cu-B物种的迁移插入启动的, 形成的手性烷基-Cu物种可对醛或酮发生亲核加成过程, 再经过硼解及氧化过程即能以高syn-选择性得到手性α-烷基-β-羟基醛或酮. 2016年, 采用类似的策略, Procter等[57]将亲电试剂拓展到亚胺, 在Cu/NHC催化下实现了联烯的不对称硼-胺烷基化反应[Scheme 18(B)]. Cao和Liao等[58]也报道了共轭二烯的不对称1,2-硼-胺烷基化反应. 另外, Hoveyda等[59]也实现了Cu(Ⅰ)/双磷配体络合物催化下丙二酸酯类缺电子烯烃作为亲电试剂和共轭二烯的不对称硼烷基化反应.
除了Cu-B催化体系, Pd催化的烯烃不对称硼碳化反应也有报道. 2019年, Engle等[60]与He等[61]分别报道了8-氨基喹啉(AQ)导向Pd催化的不对称芳基硼化反应[Scheme 19(A)和(B)]. 与Cu-B催化体系不同, 该反应经过一个类Wacker过程: 芳基亲核试剂进攻被Pd(Ⅱ)活化的烯烃, 生成Pd杂五元环中间体, 该中间体再与联硼酸酯发生转金属化, 最后经还原消除生成反式硼化产物. 由于Pd(Ⅱ)倾向于采取四配位的平面四边形结构, 而底物中烯烃与导向基AQ已经占据3个配位位点, 因而只能有1个空余位点用于手性配体配位, 使得反应对映选择性的控制相对困难. 他们最终通过使用单齿手性噁唑啉(MOX)配体实现了这一目标, 获得最高可达97%(e.e.)的对映选择性[Scheme 19(C)].

Scheme 19 Pd(Ⅱ)/L23-catalyzed asymmetric arylboration of alkenes(A), Pd(Ⅱ)/L24-catalyzed asymmetric arylboration of alkenes(B) and proposed mechanism(C)
1.3.3 烯烃的不对称硼胺化反应 基于Cu-B催化体系, 当Cu-B物种与烯烃加成后的Cu-烷基中间体被胺类亲电试剂捕获时, 则能发生硼胺化反应生成手性β-氨基硼化合物. 2013年, Hirano和Miura等[62]报道了Cu(Ⅰ)/双膦配体L8催化的苯乙烯类化合物的不对称硼胺化反应[Scheme 20(A)和(B)]. 该反应采用氧上苄基保护的羟胺衍生物作胺源, 能够以很高的syn-选择性得到苄位胺化手性硼化合物. 之后, 他们[63,64]又将烯烃进一步拓展到环内烯及硅基烯类化合物.
Scheme 20 Cu(Ⅰ) catalyzed asymmetric aminoboration of alkenes(A) and proposed mechanism(B)
2 不对称碳硼偶联反应
2.1 不对称C—H键硼化反应
虽然C—H键, 包括C(sp2)—H及C(sp3)—H键的硼化反应已经多有报道[65], 但该过程的不对称控制仍然是一个巨大挑战, 目前关于C—H键的不对称硼化反应报道较少, 且大都是基于去对称化的策略.

Scheme 21 Pd(Ⅱ)-catalyzed asymmetric C(sp3)—H borylation of cycloalkanes(A) and Ir(Ⅲ)-catalyzed asymmetric C(sp3)—H borylation of cyclopropanes(B)
2017年, Yu等[66]报道了在Pd(Ⅱ)/手性乙酰氨基噁唑啉配体L25催化下, 酰胺环烷烃的2-位C—H键能够发生去对称化硼化反应[Scheme 21(A)]. 底物中的酰胺结构具有导向作用, 可诱导Pd(Ⅱ)邻位顺式的C—H键活化过程, 生成单一的硼与酰胺处于顺式的产物. 另外, 该反应对于多种环系, 包括环丁烷、环丙烷甚至环己烷都具有良好的适用性, 能够高对映选择性地得到顺式β-硼基酰胺. 2019年, Xu等[67]使用硼基手性配体L26, 以酰胺为导向基团, 实现了Ir催化环丙烷的去对称化硼化反应, 可合成多种手性的环丙酯类化合物[Scheme 21(B)].
2017年, Shi和Hartwig等[68]报道了硅基导向的不对称C(sp3)—H硼化反应, 能够实现手性的双芳基硅基甲烷的合成[Scheme 22(A)]. 他们认为反应经历Ir(Ⅲ)-B3物种, 首先Ir(Ⅲ)-B与Si—H键复分解形成Ir—Si键, 在手性喹啉-噁唑啉配体L27的诱导下, Ir催化剂选择性地对其中一个芳环的邻位C—H键发生硼化反应, 最后再与联硼烷复分解释放出硼化产物[Scheme 22(B)].

Scheme 22 Ir(Ⅲ)-catalyzed asymmetric C(sp2)—H borylation of diarylmethylsilanes(A) and proposed mechanism(B)

Scheme 23 Ir(Ⅲ)-catalyzed asymmetric C(sp2)—H borylation of diarylmethylamines(A) and Ir(Ⅲ)-catalyzed kinetic resolution through C(sp2)—H borylation of diarylmethylamines(B)
2019年, Xu和Ke等[69]实现了Ir/手性B-N配体络合物催化的二芳基甲胺类化合物去对称化硼化反应[Scheme 23(A)], 当2个芳基不同时, 则发生动力学拆分过程, 其拆分系数(s)最高可达68[Scheme 23(B)].
2.2 不对称烯丙位硼化反应
2007年, Ito和Sawamura等[70]报道了Cu(Ⅰ)/L2催化的烯丙醇酯类化合物的γ-硼化反应[Scheme 24(A)]. 之后, Ito等[71]利用类似的催化体系实现了烯丙基缩酮的不对称γ-硼化反应[Scheme 24(B)]. 该类反应可能经历了加成-消除机理: 首先, 在酯基的导向作用下, 烯烃对Cu(Ⅰ)-B物种迁移插入生成手性烷基-Cu中间体, 接着再发生β-氧消除反应生成反式产物[Scheme 24(C)].

Scheme 24 Cu(Ⅰ)-catalyzed asymmetric borylation of allylic carbonates(A), Cu(Ⅰ)-catalyzed asymmetric borylation of (γ-alkoxyallyl)boronates(B) and proposed mechanism(C)
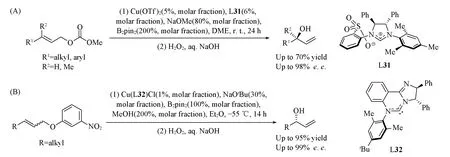
Scheme 25 Cu(Ⅰ)-catalyzed enantioselective borylation of allylic carbonates(A) and Cu(Ⅰ)-catalyzed asymmetric stereo-convergent borylation of allylic carbonates(B)
2010年, Hoveyda等[72]采用手性NHC配体L31, 结合Cu(Ⅰ)-B体系实现了烯丙醇酯类化合物的不对称γ-硼化反应. 该反应对E式或Z式及γ,γ-二取代的底物均有很好的不对称诱导效果, 且不同构型底物所得到产物的绝对构型是相反的[Scheme 25(A)]. 2011年, MacQuade等[73]使用类似的催化体系, 实现了Cu/L32催化的烯丙醇酯的所谓立体汇聚式γ-硼化反应, 即从不同Z/E构型的底物出发能
够得到相同绝对构型的产物[Scheme 25(B)]. 他们认为, 由于催化剂的特殊结构, 使其在促进烯烃对Cu-B键的迁移插入时, 对Z/E2种构型的烯烃表现出相同的面选择性, 最终给出相同绝对构型的产物.
2010年, Ito等[74]还报道了Cu/L2催化的消旋环烯丙醇类化合物的不对称γ-硼化反应, 获得了优异的收率和对映选择性(Scheme 26). 与以往的动态动力学拆分过程不同, 反应中环烯丙醇并不存在消旋化的过程, 其高收率、高选择性的实现是由于催化剂Cu-B物种对S构型与R构型的底物有着相反的面选择性识别, 从而使2种异构体最终能够转化为相同绝对构型的产物.

Scheme 26 Cu(Ⅰ)-catalyzed enantioselective borylation of racemic allylic alcohol derivatives

Scheme 27 Ni-catalyzed asymmetric C—Cl borylation(A) and proposed mechanism(B)
2.3 C—X键不对称硼化反应
除了高活性的烯丙醇类化合物, 苄基卤代物也能发生不对称的碳硼偶联反应. 2018年, Fu等[75]采用吡啶双噁唑啉配体L33与Ni(Ⅱ)的络合物作为催化剂, 实现了苄基氯代物与连频哪醇硼烷的不对称偶联反应[Scheme 27(A)]. 通常认为这类反应经历了自由基过程, 因此消旋的底物也能够完全转化得到手性的苄基硼化合物[Scheme 27(B)].
催化C—B偶联反应中还有一些比较特殊的底物类型, 2018年, Qiu和Xie等[76]报道了Pd催化的碳硼烷分子内不对称C—Br键硼化反应(Scheme 28). 该反应首先经历Pd(0)对C—Br键的氧化加成, 然后在手性单磷配体L34的诱导下, 使Pd(Ⅱ)的硼化只能发生在特定的一侧, 经还原消除即可以高对映选择性得到目标产物.

Scheme 28 Pd-catalyzed asymmetric intramolecular C—B formation of o-Carboranes
3 金属卡宾对B—H键的不对称插入反应
由于硼中心的缺电子性, 硼烷通常无法与缺电子的Fisher卡宾发生B—H键的插入反应. 但当带有孤对电子的Lewis碱配位到硼烷的空轨道之后, 可使硼中心的电子性质翻转, B—H键更具负电性, 从而使金属卡宾对硼氢的插入成为可能.
2013年, Zhu与Zhou等[77]与Curran等[78]几乎同时报道了Cu或Rh催化下的重氮酯和重氮酮衍生的卡宾对硼烷加合物的插入反应[Scheme 29(A)]. Zhu和Zhou等[79]使用手性螺环双噁唑啉配体L35, 成功实现了α-芳基重氮酯衍生的卡宾对B—H键的插入反应的不对称控制. 他们认为反应经历了如下过程: 首先铜分解重氮生成金属卡宾, 然后经过三元环过渡态的协同过程发生B—H键的插入生成有机硼化合物[Scheme 29(B)]. 该反应在B—H键插入过程中实现了反应的手性控制. Zhu等[79]还将手性螺环配体与铜的络合物作为催化剂用于重氮酮衍生卡宾的不对称B—H键插入反应, 获得了中等或较高的对映选择性.
2015年, Xu等[80]报道了Rh(Ⅲ)/手性双烯配体L36催化的稳定重氮化合物衍生的卡宾对B—H键的不对称插入反应. 除了α-芳基重氮酯, 该催化体系对α-芳基重氮酮类化合物也表现出优异的反应活性与不对称诱导效果[Scheme 30(A)]. Gouverneur等[81]系统研究了三氟甲基重氮化合物衍生的卡宾对各类杂原子-氢键的插入反应, 发现在Cu(Ⅰ)/手性双噁唑啉L37催化下, 使用三苯基膦硼烷加合物能够捕获α-三氟甲基卡宾中间体, 发生不对称B—H键插入反应, 生成相应的手性有机硼化合物[Scheme 30(B)]. 2017年, Arnold等[82]通过酶的定向进化, 实现了α-甲基重氮酯及α-芳基三氟甲基重氮化合物的不对称B—H键插入反应, 成功地用人工酶完成了生物界所不存在的C—B键形成反应[Scheme 30(C)].
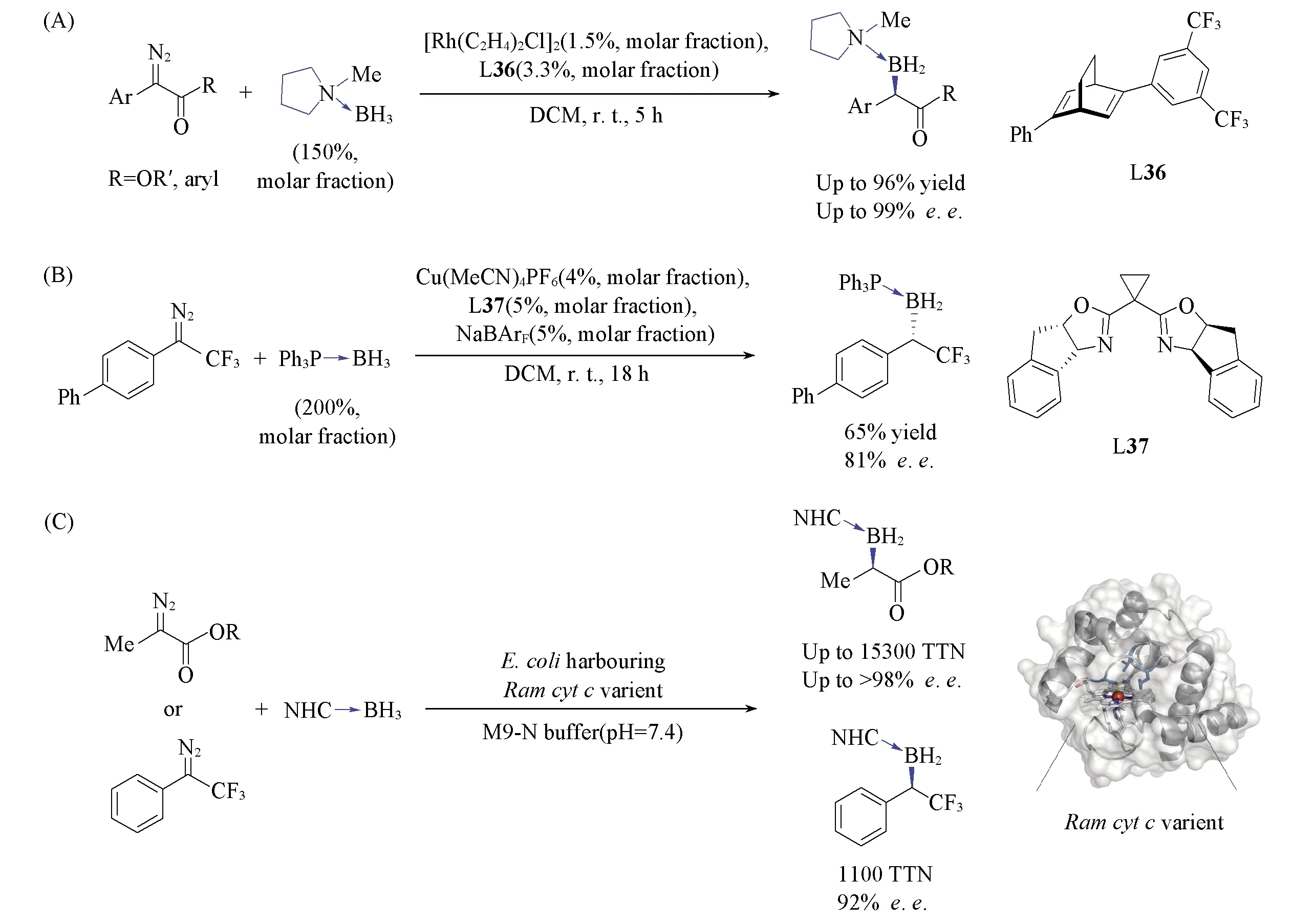
Scheme 30 Rh(Ⅲ)-catalyzed asymmetric B—H bond insertion reaction of diazoketones and diazoesters(A), Cu(Ⅰ)-catalyzed asymmetric B—H bond insertion reaction of α-trifluo methyl diazocompounds(B) and genetically programmed synthesis of chiral organoboranes(C)
除了酯或酮稳定的重氮化合物, 非稳定的芳基烷基重氮化合物对B—H键的不对称插入反应也有报道. 2018年, Zhu等[83]通过磺酰腙在强碱性及加热条件下现场生成重氮化合物的方式, 实现了手性双铑化合物C6或C7催化下的芳基烷基重氮化物对B—H键的不对称反应(Scheme 31), 并且通过两步一锅法实现了由酮到手性二级有机硼化合物的转化. 该反应将羰基官能团和硼官能团直接关联起来, 并实现了其高对映选择性转化, 具有很好的应用潜力.
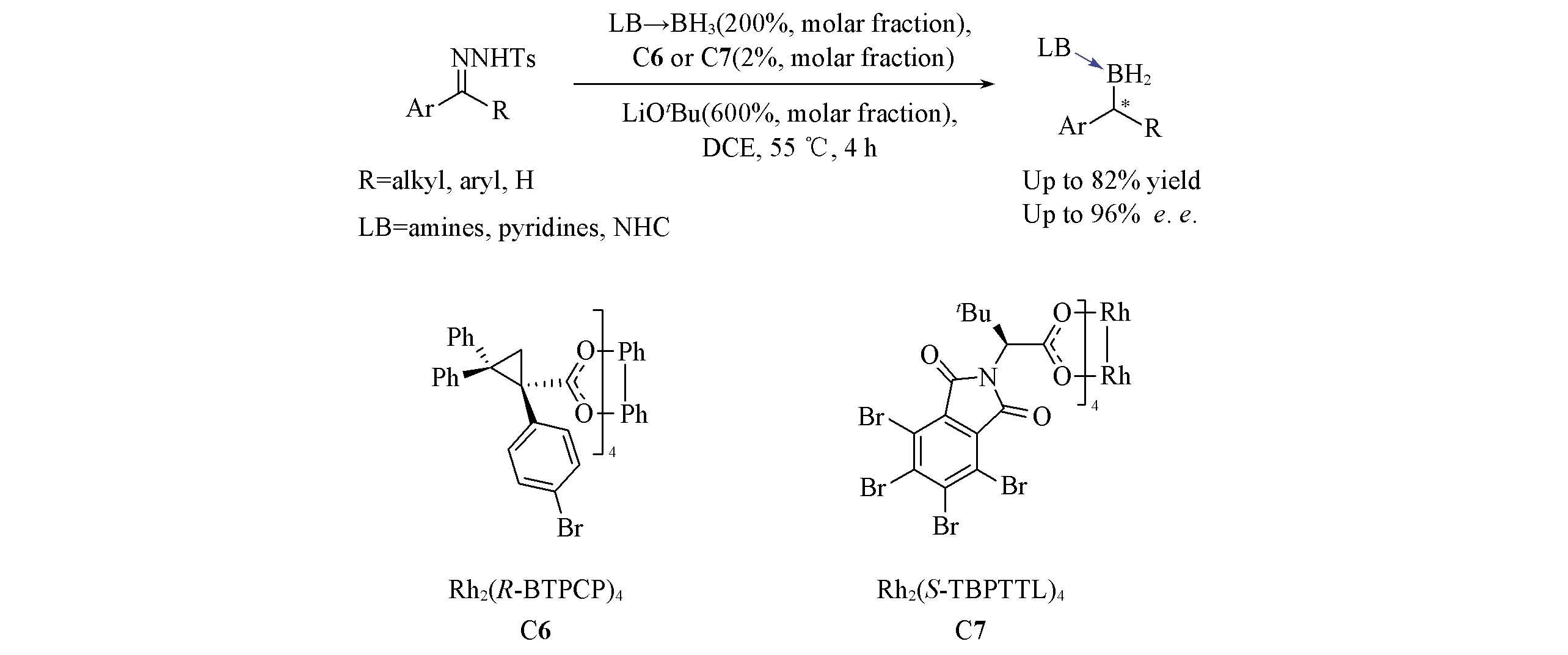
Scheme 31 Rh(Ⅱ)-catalyzed asymmetric B—H bond insertion reaction of non-stabilized diazocompounds

Scheme 32 Rh(Ⅱ)-catalyzed asymmetric B—H bond insertion reaction using carbonyl-ene-eyns as carbene precursors
除了重氮化合物, 更加稳定易得的炔烃也能在金属催化下生成金属卡宾进而发生硼氢插入反应. 2017年, Zhu和Zhou等[84]报道了手性双铑络合物C6或C7催化下羰基-烯-炔类化合物的不对称硼氢插入反应(Scheme 32). 炔基在Rh(Ⅱ)的活化下能够被羰基分子内进攻, 经环化重排得到2-呋喃基卡宾, 其对B—H键发生不对称插入反应即可得到双芳基硼基化合物. 产物中的硼基团还可以转化为各类硼酸酯以及直接转化为醇, 且在这些转化过程中几乎完全保持了对映选择性. 最近, Zhu和Zhou等[85]还实现了金催化端炔的氧化硼化反应, 其关键步骤也涉及金属卡宾对硼烷加合物B—H键的插入反应; 该反应为α-羰基硼化合物的合成提供了新方法, 但其不对称转化尚未见报道.
4 其它类型的不对称碳硼成键反应
2015年, Toste等[86]报道了Pd与手性磷酸C8共催化的端烯、芳基重氮盐与联硼烷的1,1-芳基硼化反应, 能够一步得到苄位手性的二级硼酸酯[Scheme 33(A)]. 与一般配体诱导的不对称控制模式不同, 反应中的手性诱导产生于手性磷酸负离子, 其与芳基Pd(Ⅱ)正离子形成的离子对与烯烃迁移插入形成烷基Pd中间体, 该中间体经还原消除再重新插入后形成更加稳定的Pd位于苄位的中间体, 随后其与联硼酸酯发生转金属化再还原消除得到产物[Scheme 33(B)].

Scheme 33 Pd(Ⅱ)-catalyzed asymmetric 1,1-arylboration of alkenes(A) and proposed mechanism(B)
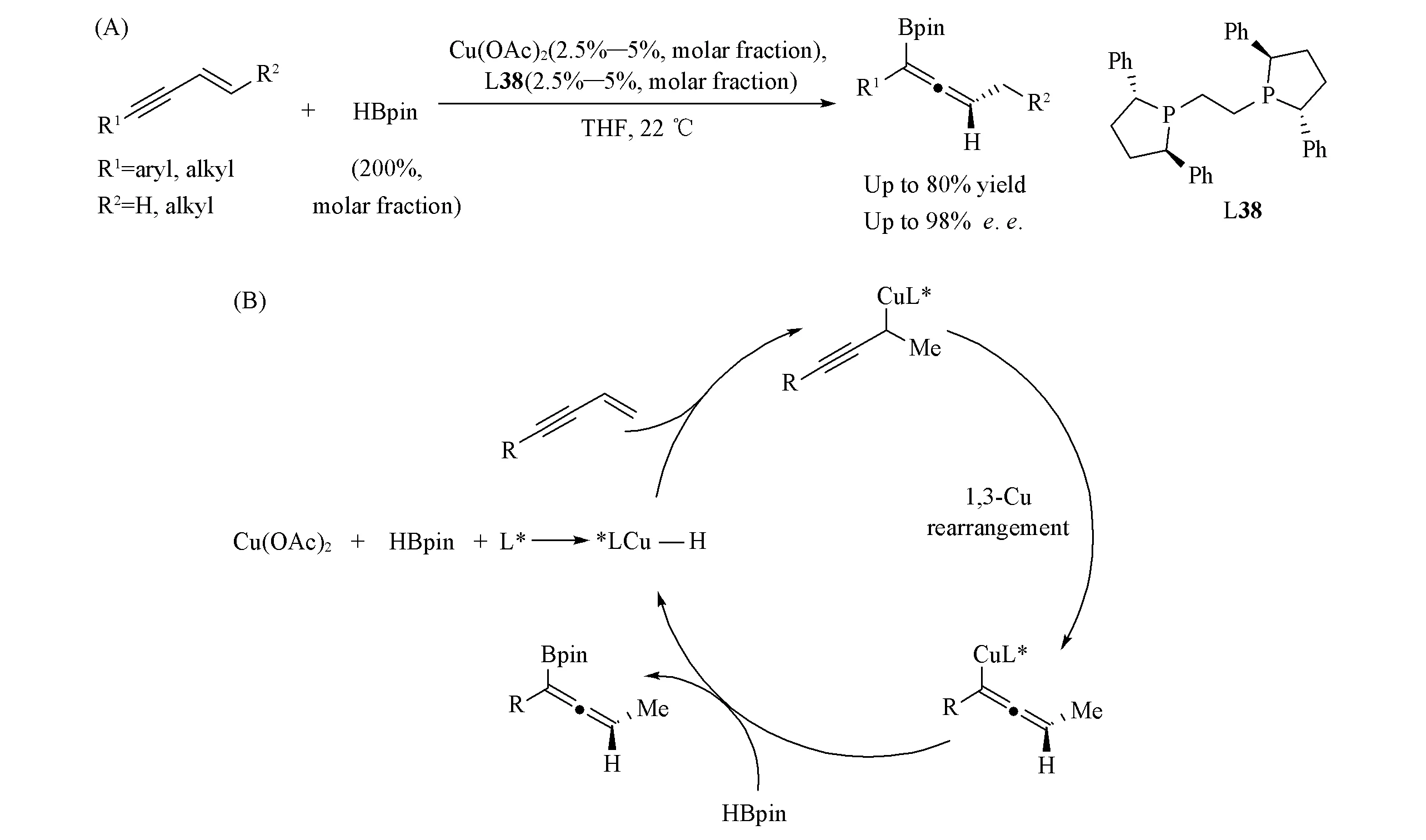
Scheme 34 Cu(Ⅰ)-catalyzed asymmetric rearrangement/hydroboration for the synthesis of allenyl organoboranes(A) and proposed mechanism(B)
2018年, Hoveyda等[87]报道了Cu/手性双磷配体L38催化的1,3-烯炔不对称重排/硼氢化反应, 高对映选择性地合成了轴手性联烯硼化合物[Scheme 34(A)]. 他们认为, 反应首先由Cu—H物种对烯烃的加成生成炔丙基Cu中间体, 随后Cu发生1,3-迁移生成联烯Cu物种, 这一步是反应的手性控制步骤, 随后联烯铜物种硼解即可生成产物, 同时再生催化剂[Scheme 34(B)]. Ge等[88]和Engle等[89]也分别报道了基于Cu—H物种的类似反应.
5 总结与展望
原则上, 只要碳硼成键反应的最终产物具有手性, 就可能通过在催化过程中引入手性催化剂对反应的对映选择性加以调控. 本文从反应类型上综合评述了不对称催化条件下构筑C—B键的方法, 可以看到, 得益于手性催化剂的引入, 多种类型的手性有机硼化合物能够被高效合成. 总体而言, 目前报道的手性有机硼的类型大多局限于碳中心手性, 而关于轴手性、面手性及硼中心手性的手性有机硼化合物催化合成的报道很少, 限制了有机硼化合物的应用范围, 因此发展结构多样性手性有机硼化合物的合成方法对有机硼化学的发展具有重要意义. 从结构多样性合成的角度来看, 目前发展的各类不对称碳硼成键方法都有其局限. 硼试剂对烯烃的加成反应是合成手性烷基硼的重要方法, 然而其底物类型较为局限, 因此发展更高效的手性催化剂, 实现非活化的多取代烯烃、非对称双烷基或双芳基烯烃的区域选择性与对映选择性精准调控具有重要意义. 基于C—H键活化的不对称C—B键偶联反应在官能团转化中具有独特优势, 但由简单底物出发的不对称C—B键偶联反应仍然是巨大的挑战, 发展更为高效的C—H键的活化方式, 特别是C(sp3)—H键-活化方式, 并实现其手性控制, 无疑是该类反应的发展方向. 卡宾对B—H键插入反应的出现有望将卡宾结构多样性的优势引入到有机硼的合成中, 拓展更多类型卡宾对B—H键的插入反应, 实现其选择性有效调控, 将推动该反应的实际应用. 另外, 在碳碳成键反应中引入碳硼成键, 也是达成结构多样性有机硼合成的有效方法, 此方面的研究同样值得期待[90,91].
