脊髓小脑性共济失调3型1例报告及其相关文献复习
2020-07-07徐凯翎张艺凡陈美秋
徐凯翎, 张艺凡, 楚 兰, 陈美秋, 夏 聪, 罗 婷, 刘 磊
脊髓小脑性共济失调(Spinocerebellar ataxia,SCA)是一组由小脑及其传入和传出连接变性引起的常染色体显性共济失调疾病,主要临床表现包括姿势和步态的异常、构音障碍、眼球运动障碍、视网膜病变、锥体外系及锥体束病变、周围神经损害等[1]。SCA属于罕见病,在欧洲不同地区进行的流行病学研究发现患病率为(0.9~3.0)/10万人[2]。根据临床表现或遗传学分类,迄今为止已经确立了40种SCA,其中包括SCA1-40。鉴定了28种SCA的致病基因[3],本文报道1例确诊为脊髓小脑性共济失调3型(Spinocerebellar ataxia type 3,SCA3)病例的临床表现、病史及相关文献报道如下。
1 临床资料
患者,女,49岁,主因“行走不稳3 y,言语含糊1 y,加重1 w”于2019年11月12日入住我院。3 y前患者无明显诱因出现行走不稳,主要表现为行走时左右摇晃,步基宽,偶有跌倒,但仍可独立行走,无头晕、头痛、意识障碍;无言语含混、饮水呛咳、吞咽困难;无发热、恶心、呕吐,予以相关药物治疗(具体不详)后未见明显好转。1 y前患者开始出现言语含糊,表现为吐字不清、发音困难,伴饮水呛咳,未系统诊治。1 w前患者上述症状较前加重,就诊当地乡镇医院予“天麻素、血塞通”药物治疗后,饮水呛咳较前缓解,但行走不稳及言语含糊仍呈进行性加重,为求进一步诊治遂就诊我院。发病以来精神、饮食欠佳,二便正常,睡眠较差,表现为入睡困难,体重未见明显增减。入院时否认糖尿病、高血压、冠心病等病史;否认烟酒等不良嗜好。家族史:父母已逝(因年老去世),有2哥1姐1妹,大哥已故,大姐完善头部磁共振示小脑萎缩,无相关临床表现,余兄弟姐妹体健。婚育史:育有1儿1女,均体健;个人史无特殊。
查体:内科检查:发育正常,营养中等,查体合作,卧位血压:120/94 mmHg,站立位血压:100/70 mmHg。神经系统检查:神志清楚,言语含糊,构音障碍,查体合作。双眼球各向活动可,有水平眼震。双侧咽反射迟钝。下肢腱反射(+),步基宽。双上肢轮替、指鼻、指指试验欠稳准,双下肢跟膝胫试验欠稳准,一字步欠稳准,Romberg征(+)。左下肢Babinski征(+)、Chaddock征(+)、Oppenheim征(+);右下肢Babinski征中性。实验室检查:乙肝5项(定量)、血常规、甲状腺功能、心肌标志物、肝肾功、电解质、血脂、心肌酶、传染病初筛、凝血全套、免疫7项、血清维生素D水平未见明显异常。头部MR平扫:(1)符合橄榄-桥脑-小脑萎缩(OPCA)MR征象(见图1A)。头部MR增强扫描:符合橄榄-桥脑-小脑萎缩(OPCA)MR征象。颈椎MR平扫:颈段脊髓萎缩(见图1B)。胸椎MR平扫:胸段脊髓萎缩并中央管扩张(见图1C)。头部DWI、SWI未见明显异常。头部MRA:右侧椎动脉V5段纤细,考虑发育所致。神经传导检查、肌电图(肛门括约肌)检查:(1)周围神经损害;(2)肛门括约肌EMG呈神经源性损害。植物神经功能检查:SSR:双上肢潜伏期延长、双下肢未引出肯定波形,提示:四肢SSR异常。周围神经结果:(1)双侧正中神经轻度损害;(2)双侧胫神经、双侧胫后神经、双侧腓总神经、左侧坐骨神经损害。ATXN3基因检测“CAG重复序列杂合突变,数目分别为14和71”(见图2);其女ATXN3基因检测“CAG重复序列杂合突变,数目分别为27和72”(见图3);患者家系图谱分析(见图4)。
结合患者病史、体征、辅查及基因检测结果,临床考虑诊断:脊髓小脑性共济失调3型,入院予以脑苷肌肽、桂哌齐特改善循环,艾地苯醌改善睡眠,语言训练、吞咽训练、姿势训练,以改善语言、吞咽、平衡功能,出院时患者睡眠障碍较前好转,言语含混、饮水呛咳、行走不稳未见明显改善。

图1 A.颅脑矢状位MRI T1WI;B.颈椎矢状位MRI;C胸椎矢状位MRI
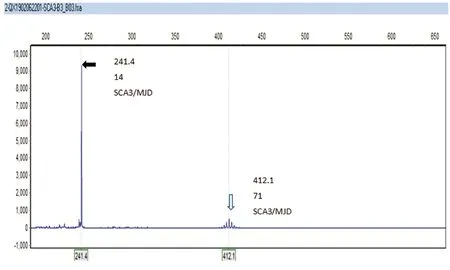
图2 STR分析显示患者SCA3/MJD基因CAG重复数目,正常等位基因重复14次(黑色箭头所示),异常等位基因重复71次(白色箭头所示)

图3 STR分析显示患者之女SCA3/MJD基因CAG重复数目,正常等位基因重复27次(黑色箭头所示),异常等位基因重复72次(白色箭头所示)
图4 本例患者脊髓小脑性共济失调3型三代家系图
2 讨 论
脊髓小脑性共济失调3型(Spinocerebellar ataxia type 3,SCA3)也称为马查多-约瑟夫病(Machado-Joseph disease,MJD),首次报道于1970年来自William Machado和Antone Joseph这两个家族,因此命名为Machado-Joseph Disease,是全世界最常见的SCA[4]。致病基因ATXN3定位于染色体14q32.1,编码致病蛋白ataxin-3/MJDp。基因突变是CAG在染色体14q24.3-q32.1上重复扩增。正常人的CAG重复数范围为10~44次,而SCA3患者的CAG重复大小范围为61~87次。通常,疾病严重程度与CAG重复扩展的程度相关,CAG重复次数越多,疾病的严重程度越高,发病越早[5]。本例患者及其女儿完善基因检测显示SCA3 相关基因的CAG重复数分别为71次和72次,符合SCA3的基因突变特征。
2.1 临床特点 SCA3具有高度的遗传异质性和临床变异性,反映了中枢神经系统细胞变性的模式-多个神经元系统可受到影响,包括小脑、脑干、颅神经、基底节和脊髓[6]。SCA3的症状发作通常始于成年后第三至第五个十年,并随着年龄的增长而缓慢发展,主要核心表现为小脑共济失调、眼外肌麻痹、突眼征、凝视诱发眼球震颤、面舌肌肌束颤动、构音障碍、吞咽困难、饮水呛咳以及不同程度的锥体系和锥体外系症状和周围神经病变[7],其他非运动症状包括睡眠、认知及精神障碍等[8]。尽管SCA3患者的病情严重程度和进展速度各不相同,但脑干相关功能的衰竭通常会导致症状发作并于症状发生后10~15 y内死亡[9]。磁共振成像研究最常检测到基底神经节的异常,脑桥以及小脑上脚、小脑下脚萎缩以及第四脑室扩张。本例患者慢性起病,3 y前曾出现双下肢行走不稳,期间治疗后临床症状未见明显缓解。1 y前走路不稳症状加重,并出现言语含混,完善EMG示四肢神经源性损害,头部MRI示橄榄-桥脑-小脑萎缩。颈椎MR示:颈段脊髓萎缩。胸椎MR示:胸段脊髓萎缩并中央管扩张。结合病史及临床表现,基因检测结果明确了SCA3的诊断。患者家系调查中患者直系家系3代共9人 ,第一代患病情况不详,其余两代共1例疑似、2例确诊SCA患者,先证者父母及兄弟姐妹因故未进行基因检测仅根据病史诊断;先证者大姐目前完善头部磁共振示小脑萎缩,但目前尚未出现相关临床表现,考虑疑似病例。先证者女儿完善基因检测后确诊,目前尚未出现临床表现,告知其应动态复查基因检测并进行治疗以延缓疾病进展程度。该患者家族史阴性,未能检测直系亲属的基因,推测该例患者可能是散发的SCA3。本例患者发病年龄较晚,进展迅速,考虑和SCA3临床表现变异大有关系。
2.2 治疗 目前尚无有效的SCA治疗方法,目前的治疗主要是延缓疾病的进展,对症治疗(包括改善共济失调、减轻疼痛、缓解肌痉挛等)及康复治疗(包括物理治疗、言语治疗和职业治疗)[10]。随着近年来分子生物技术的迅速进展及对疾病发病机制的深入研究,目前能够使用干扰基因表达的技术,如RNA干扰,寡聚反义核苷酸、基因治疗及基于表观遗传学的治疗,但上述疗法尚处于I期临床试验[11],相信在不久的将来,可找到更为可靠的方法对SCAs进行有效的预防和治疗。
