肉芽肿性多血管炎合并弥漫性肺泡出血一例报道并文献复习
2020-04-27陆翠苏禹同滕佳临杨程德叶俊娜
陆翠,苏禹同,滕佳临,杨程德,叶俊娜*
本文价值:
弥漫性肺泡出血是肉芽肿性多血管炎的罕见表现,表现为咳嗽、呼吸困难和咯血等,一旦发生,进展迅速,死亡率高。本文报道1例肉芽肿性多血管炎合并弥漫性肺泡出血患者,早期诊断后即给予积极治疗,预后良好。提示对此类患者早期识别和积极抢救对预后至关重要。
肉芽肿性多血管炎(GPA)曾被称为韦格纳肉芽肿(WG),是一种少见的全身性疾病,其特征为坏死性肉芽肿性血管炎,主要影响上呼吸道、肺部和肾脏[1]。弥漫性肺泡出血(DAH)是GPA的罕见表现,一旦出现,病情危重,死亡率高,目前仅有少数个案报道[2-12]。本研究报道上海交通大学医学院附属瑞金医院风湿免疫科收治的1例GPA合并DAH患者,并结合相关国内外文献,分析该病的病因、临床表现、实验室及影像学特征、治疗和预后,以期提高临床医生对该病的认识。
1 病例简介
患者,女,24岁,因鼻腔血性分泌物3个月、咳嗽1个月于2018-10-11入本院。患者2018年7月开始出现反复鼻腔血性分泌物,外院鼻窦CT提示鼻窦炎,给予抗感染等治疗,效果不佳。1个月前出现咳嗽、痰中带血,伴胸痛、听力下降,2周前出现反复结膜充血,伴双下肢麻木、疼痛不适,病程中发热2次,肉眼血尿1次,无呼吸困难、咯血,无皮疹、关节肿痛。有重症肌无力病史。查体:神清,气平,持续性咳嗽,双眼球结膜充血(见图1),鼻腔血性分泌物,上颌窦压痛(+),双肺呼吸音低,未闻及啰音。辅助检查:白细胞计数(WBC)9×109/L,血红蛋白(Hb)108 g/L,血小板计数(PLT)308×109/L;红细胞沉降率(ESR)74 mm/1 h;尿常规:蛋白(+),红细胞31~50个/HPF,隐血(+++);24 h尿蛋白定量326 mg;抗中性粒细胞胞质抗体(c-ANCA)阳性,蛋白酶3特异性抗中性粒细胞胞质抗体(PR3-ANCA)209.90(参考范围<20.00),抗中性粒细胞核周抗体(p-ANCA)阴性,髓过氧化物酶抗体(MPO)阴性;血清清蛋白26 g/L;血肌酐处于参考范围内;抗核抗体(ANA)阴性、抗双链DNA(dsDNA)抗体阴性、抗可溶性抗原抗体(ENA)阴性;类风湿因子(RF)阴性;补体处于参考范围内。胸部CT:双肺多发斑片、结节及索条影伴实变影;左肺门饱满、右肺门增大,两侧主气管及部分支气管管腔变窄、管壁增厚(见图2A);肌电图:左侧正中神经传导延迟。诊断:GPA,因患者剧烈咳嗽、胸部CT较2周前新发弥漫性斑片状阴影,考虑合并DAH,予甲泼尼龙500 mg冲击治疗3 d,逐渐减量,并静脉注射人丙种球蛋白(IVIG)20 g 5 d,环磷酰胺(CTX)0.4 g冲击治疗1次,第5天复查胸部CT双肺斑片状磨玻璃影、实变影明显好转(见图2B),第12天复查胸部CT病灶进一步被吸收(见图2C),1个月后复查c-ANCA阴性,PR3-ANCA 136.64,随访半年,病情稳定,CTX冲击治疗共7次,未再出现DAH。
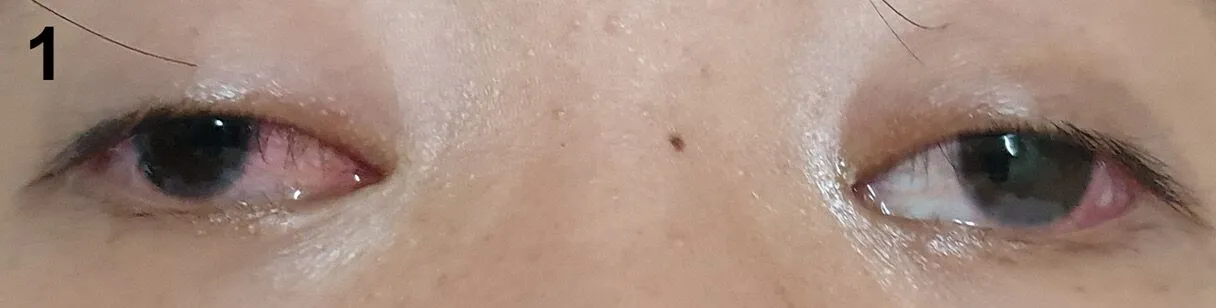
图1 双眼球结膜充血Figure 1 Binocular conjunctival congestion
2 文献检索
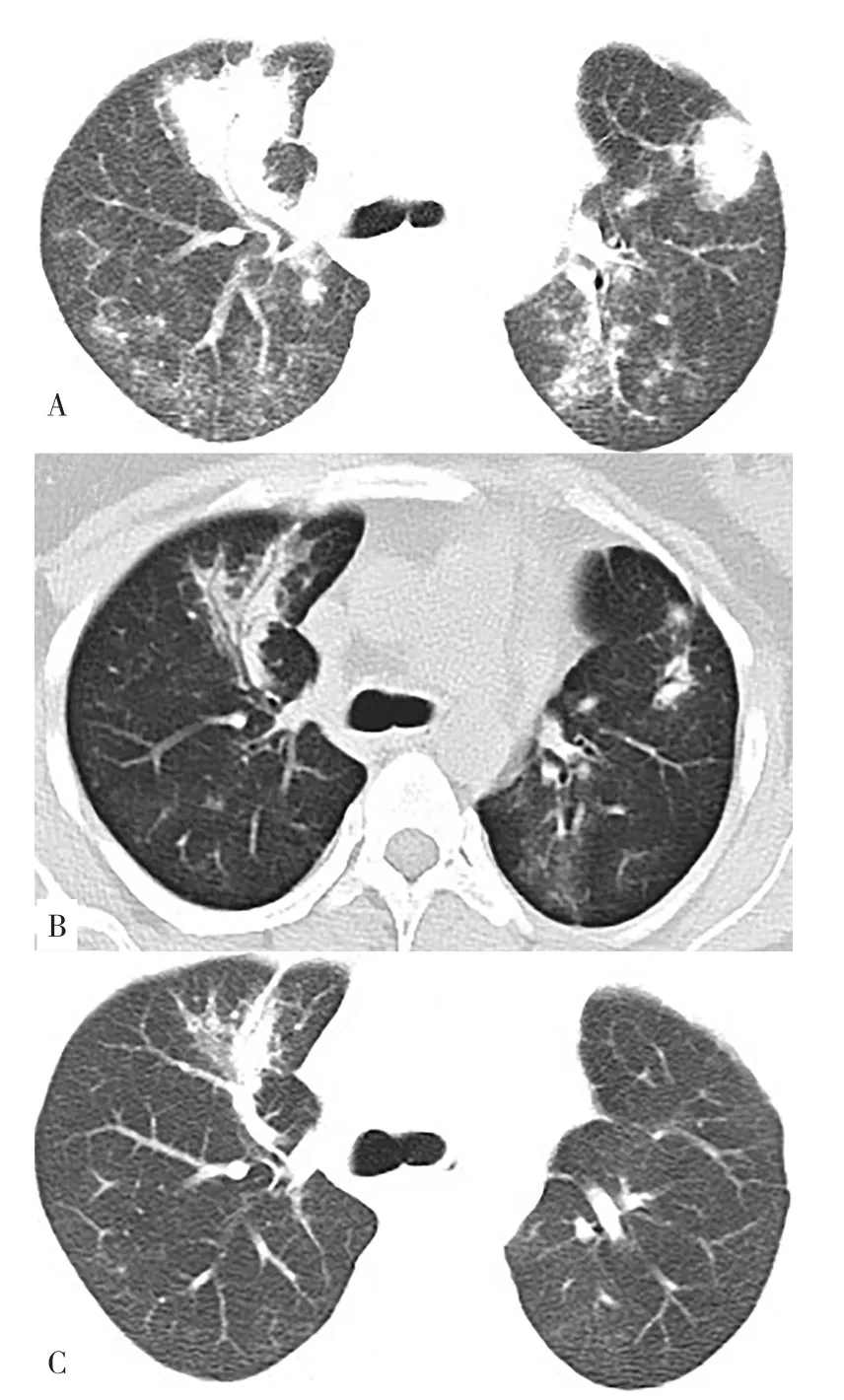
图2 胸部CT检查结果Figure 2 The results of chest computed tomography
以“韦格纳,血管炎,肉芽肿性多血管炎,肺出血,肺泡出血”为关键字,搜索万方数据知识服务平台、中国知网、PubMed数据库1980年1月—2019年2月公开发表的GPA/WG合并DAH的病例。纳入标准:有具体病例资料,诊断明确。排除标准:重复报道、同时合并其他结缔组织病、荟萃分析和综述。
共收集到国内外文献报道13例,剔除同时合并系统性红斑狼疮(SLE)1例,共12例[2-12],结合本病例,共纳入13例患者。其中男7例,女6例;年龄7~70岁,平均年龄(38.6±19.3)岁;病程1 d~6个月,平均病程(72.1±62.7)d,病程较短,多数在发病3个月内发生DAH;治疗多选择大剂量激素冲击治疗和CTX治疗;死亡率23.1%(3/13);详见表1。
13例患者中,10例患者有Hb记录,其Hb为(75.8±24.9)g/L;多数患者伴发热,合并肾脏、眼、耳、鼻等多器官受累,常见咳嗽(83.3%)、呼吸困难(83.3%)、咯血(41.7%)等表现,胸部影像学检查多见多发斑片、磨玻璃影或弥漫性浸润影、结节、实变影等(见表2)。1例行右上肺叶切除术,病理示肺泡壁弥漫性增厚,含中等量单核细胞,偶见嗜酸粒细胞;肺泡含有大量巨噬细胞(其中一些含有含铁血黄素);小范围组织性肺炎并坏死[2]。1例于就诊当天死亡,尸检肺部病理示:肺弥漫性水肿,左肺上叶结节,弥漫性肺泡损伤及DAH,出血区域见毛细血管炎,肺泡间隔被中性粒细胞破坏[6]。
3 讨论
GPA是一种与抗中性粒细胞胞质抗体(ANCA)相关的血管炎,主要累及小血管,其特征是坏死性肉芽肿性炎症,临床表现和预后因不同的受累器官及系统而有所差异,发病初期可表现为非特异性全身症状,以后可累及重要脏器,威胁生命。DAH是肺血管炎的严重表现,最常见于小血管炎,特别是GPA和显微镜下多血管炎(MPA),也可见于Goodpasture综合征、过敏性紫癜和SLE,少见继发于感染和药物或毒素[13]。GPA合并DAH临床较为罕见,一旦出现多急性起病,病情进展迅速,死亡率高。本研究报道1例GPA合并DAH患者,结合相关国内外文献,分析该病的病因、临床特点、治疗和预后,以期提高临床医生对该病的认识。本例患者经早期诊断及积极救治,预后良好,因此提高对该病的认识、早期积极治疗对该病的预后至关重要。
3.1 流行病学 虽然肺是GPA中最常见的靶器官,但出现DAH者较为罕见,发生率仅为6.1%~14.9%[14-15]。本研究发现该病平均年龄40岁左右,儿童及老年人较少见,男女比无明显差异。
3.2 病因和发病机制 ANCA可诱导中性粒细胞脱颗粒,产生氧自由基和释放蛋白酶,导致肺血管内皮和毛细血管损伤,引起小血管弥漫性肺泡内出血[16],这可能是GPA患者发生DAH的原因。
3.3 临床表现 本研究文献分析显示,DAH多在GPA起病3个月内出现,前期可有发热,同时伴结膜炎、鼻塞、鼻腔血性分泌物、中耳炎、皮疹、关节痛、肾脏损害等其他系统表现,其中以肾脏损害(83.3%)最常见。SAUVAGET等[17]曾报道66例GPA,其中合并DAH的9例均伴有急进性肾炎。
本研究文献分析显示,肺部表现主要为咳嗽(83.3%)、呼吸困难(83.3%)、咳痰(41.7%)、咯血(41.7%)、胸痛(25.0%)和痰中带血(25.0%),多数有严重呼吸困难,需气管插管等辅助通气支持。本例患者表现为呼吸困难及咳嗽,咳嗽为难以控制的刺激性咳嗽,并痰中带血,但无咯血。有研究认为约1/3的DAH患者不会出现咯血,因为肺泡体积足以容纳血液并阻止其扩散到支气管中[18]。
DAH可作为GPA患者的首发表现,也可在其他系统表现之后出现或在糖皮质激素和免疫抑制治疗期间出现,甚至有报道显示在大剂量激素冲击治疗后出现DAH[3-4,19],DAH经治疗好转后仍有复发可能[11,19],因此不论在疾病的任何阶段均需要警惕DAH的发生。
3.4 实验室检查 本研究文献分析显示,GPA患者血常规多见轻到中度贫血,提示短期内出现不明原因的Hb下降需特别注意DAH的发生。尿液检查可见蛋白尿、血尿,少见尿管型,可伴24 h尿蛋白定量升高。ESR多增快,提示DAH多在疾病活动时发生。所有患者有c-ANCA或PR3阳性,病情好转后可转阴或滴度下降[11],本例患者经治疗好转,1个月后复查,c-ANCA转阴,PR3滴度明显下降。因此,c-ANCA或PR3可作为疾病治疗后疗效判定的指标之一。部分GPA患者可有血肌酐升高、血清清蛋白下降,肝功能多正常。ANA、抗肾小球基底膜抗体(GMB)、MPO、p-ANCA阴性可与其他弥漫性结缔组织病及小血管炎鉴别。
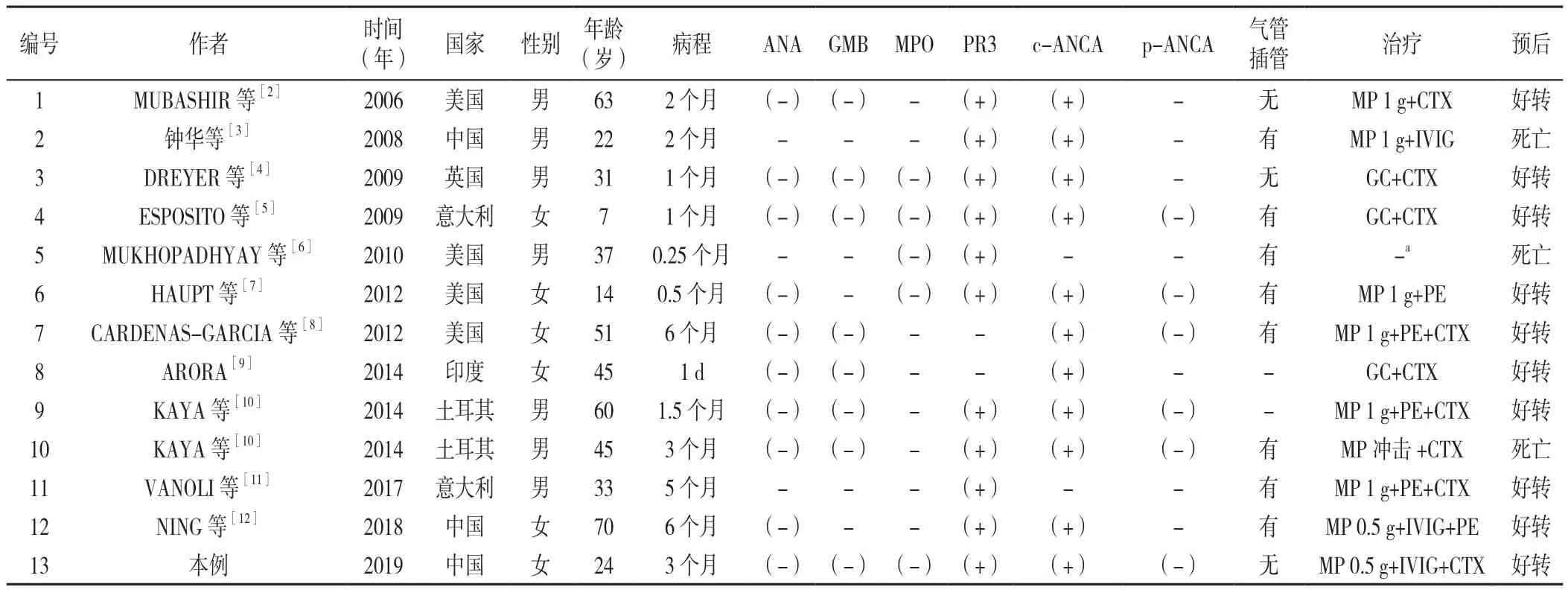
表1 GPA合并DAH患者基本情况Table 1 The general information of granulomatosis with polyangiitis patients with diffuse alveolar hemorrhage
3.5 胸部影像学检查 胸部影像学检查在GPA合并DAH诊断中具有重要作用。本研究文献分析显示,胸部X线片或胸部CT最常见的影像学表现为双肺多发斑片状、磨玻璃影或弥漫性浸润影(84.6%),局部实变影(69.2%),部分可及支气管空气征、胸腔积液,少见白肺(7.7%),GPA特异的结节样病灶仅见于38.5%的患者;关于肺部结节的检出率,胸部CT明显优于胸部X线片。治疗后肺部影像可在短期内明显好转,NING等[12]报道1例患者治疗第2天复查胸部X线片即明显好转,而本例患者治疗4天后肺部CT复查病灶明显吸收。有时DAH与肺部感染、急性肺水肿较难区分,需注意鉴别。
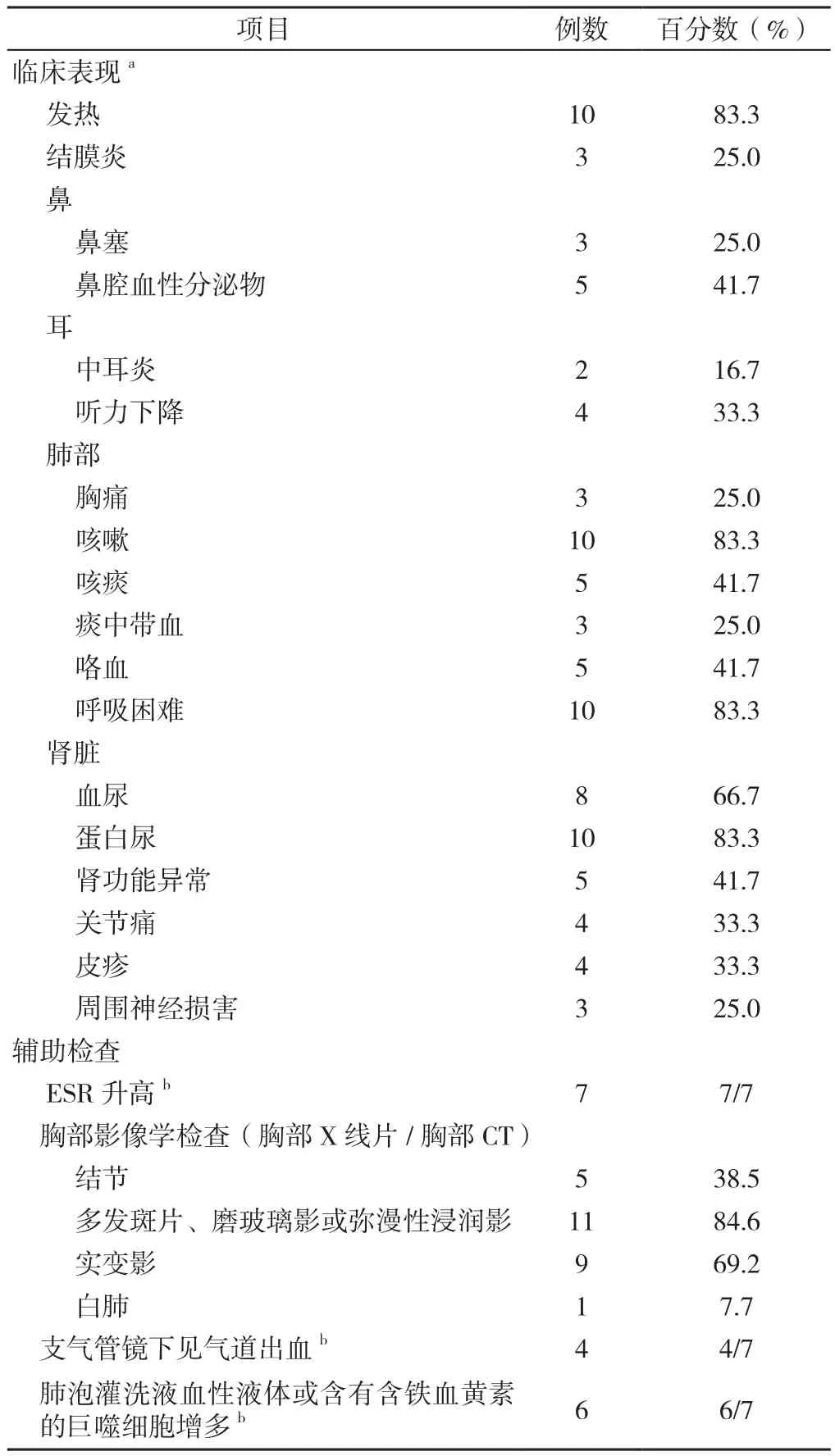
表2 GPA合并DAH患者临床表现及辅助检查结果Table 2 Clinical characteristics,laboratory and chest roentgenogram features of granulomatosis with polyangiitis patients with diffuse alveolar hemorrhage
3.6 支气管镜 支气管镜和肺泡灌洗液在确诊DAH中占重要地位,本研究文献分析显示,部分GPA患者可在支气管镜下直接见到气道出血(57.1%),而多节段肺泡灌洗液呈血性或肺泡巨噬细胞含有含铁血黄素亦可确诊DAH,支气管镜下还可同时进行细胞分析、肺活检除外感染、肿瘤。
3.7 病理 因取材困难,目前的病理研究资料较为有限。本研究文献分析显示,肺毛细血管炎为DAH主要的病理表现,可见肺泡壁弥漫性增厚,含中等量单核细胞,偶见嗜酸粒细胞;肺泡结构被中性粒细胞破坏;肺泡内可见含有含铁血黄素的巨噬细胞[2,6]。
3.8 治疗和预后 本研究文献分析显示,GPA治疗均选用激素,多选择大剂量激素冲击(0.5~1.0 g/d)治疗约3 d后逐渐减量,同时进行CTX冲击治疗和/或血浆置换、IVIG等。也有少数患者选择利妥昔单抗(RTX),VANOLI等[11]报道1例GAP合并DAH,经上述方案治疗好转2个月后再次复发,给予激素冲击和RTX治疗后再次缓解。GPA进展迅速,死亡率高达23%~60%[20],预后不良,诊断明确后需尽早进行激素冲击治疗,本例患者入院当天即给予大剂量激素冲击治疗,预后较好,临床如不能及时做出准确判断,大量肺泡出血后多数继发感染,将极大增加治疗难度。多数GPA合并DAH患者有严重呼吸困难,需气管插管辅助通气,部分患者在气管插管时吸出大量鲜红色血性液体,可帮助明确DAH诊断,而需要机械通气可能预示了预后不良[21];除呼吸系统损害外,合并有其他重要脏器损害(常见肾功能衰竭)、继发难以控制的感染也是GPA患者预后不良的影响因素。接受免疫抑制治疗的患者感染风险高,而治疗原发病免疫抑制剂为基础用药,在如何恰当选择免疫抑制剂治疗原发病和预防感染的发生之间掌握平衡一直是风湿科医生面临的一项重要挑战。有数据表明,用甲氧苄啶-磺胺甲基异恶唑(TMP-SMX)治疗可能有助于降低GPA患者缓解后的复发率[22],本研究中共有38.5%(5/13)的患者(包括本例患者)接受了TMP-SMX治疗[2,4,8,12],5例患者在随访中均长期存活,因此 GPA 合并DAH患者使用TMP-SMX可能能改善预后。
总之,DAH是GPA的一种严重临床综合征,临床表现为咳嗽、呼吸困难、咯血等,同时合并肾功能损害的患者发生DAH风险较高,短期内不明原因的Hb下降、肺部影像学检查显示弥漫性浸润影和实变影提示DAH可能,支气管镜和肺泡灌洗液见血性液体及含有含铁血黄素的巨噬细胞可明确诊断。其治疗多选择大剂量激素冲击治疗、CTX和血浆置换等。GPA一旦发生DAH,多病情危重,威胁生命,早期诊断和及时治疗可改善预后。
作者贡献:陆翠进行文章的构思与设计、文献/资料收集和整理,并撰写论文;苏禹同、滕佳临、叶俊娜进行文章的可行性分析;杨程德、叶俊娜进行论文的修订;叶俊娜进行英文的修订;陆翠、叶俊娜对文章整体负责,监督管理。
本文无利益冲突。
