血清载脂蛋白C3在胰岛素抵抗疾病中的研究进展
2020-04-18杨滢谭秋晓李荔陈志静
杨滢 谭秋晓 李荔 陈志静
1广东省妇幼保健院暨广州医科大学附属广东省妇儿医院妇科(广州510010);2中山大学附属第七医院妇科(广东深圳518107)
血清载脂蛋白C(apolipoprotein⁃C,ApoC)是载脂蛋白家族中的一员。APOC中,以ApoC3浓度最高,其对稳定脂蛋白结构、结合和转运脂质、调节脂蛋白代谢都有重要作用[1]。ApoC3是一种小分子糖蛋白,主要由肝脏合成,并进入血液循环。它是富含三酰甘油脂蛋白的主要成分,也是高密度脂蛋白的次要成分。胰岛素抵抗(insulin resis⁃tance,IR)是指胰岛素作用对的靶器官和靶组织(主要是肝脏、肌肉和脂肪组织)对胰岛素生物作用的敏感性降低,是多种代谢异常的基础[2]。目前,有关研究[3⁃7]表明,在IR状态下,如部分多囊卵巢综合征(polycystic ovary syndrome,PCOS)、2型糖尿病(diabetes mellitus type 2,T2DM)及动脉粥样硬化(atherosclerosis,AS),肝脏和肠道表达和分泌的ApoC3增多,提示ApoC3与IR有密切关系,参与IR的发生与发展,并可作为新的治疗靶点。ApoC3在IR相关疾病中的作用及机制也成为了当今研究热点之一,现对此作一综述。
1 ApoC3与IR发病机制
1.1 ApoC3导致肝脏IR ApoC3水平的升高可导致肝脏IR的发生。高脂饮食干预的ApoC3过表达小鼠,肝脏二酰基甘油含量增加,蛋白激酶C⁃ε活化和胰岛素刺激的丝氨酸⁃苏氨酸蛋白激酶2活性降低,血浆胰岛素升高,胰岛素敏感性下降,最终发展为肝脏IR[3]。人群研究也显示ApoC3高表达与IR发生密切相关,研究者发现与对照组比较,IR的患者血清ApoC3水平升高[8]。
1.2 ApoC3通过抑制脂蛋白脂肪酶导致IR脂蛋白脂肪酶(lipoprotein lipase,LPL)是溶酶体中的一种水解酶,在血管内皮表面发生作用,帮助细胞摄取脂肪酸。在小鼠骨骼肌细胞中过表达LPL[9],其葡萄糖耐量降低,血糖升高;且骨骼肌细胞对胰岛素刺激不敏感,骨骼肌细胞葡萄糖摄取、糖酵解和糖原合成减少。相反,在小鼠骨骼肌细胞敲除LPL后,由于摄取游离脂肪酸功能受到抑制,其胰岛素敏感性升高[10]。细胞内磷脂酰肌醇3⁃激酶(phosphatidylinositol 3⁃kinase,PI3K)/蛋白激酶B(protein kinase B,AKT)/过氧化物酶体增殖物激活受体通路激活,可导致LPL表达升高,进而影响脂质积累过程[11]。
具有肌肉和肝脏特异性过表达LPL的转基因小鼠的研究[12]发现,肝内脂肪酸衍生代谢物的积累导致糖原合酶的胰岛素活化受抑制,使得胰岛素刺激的肝糖原合成减少和抑制肝脏中内源性葡萄糖产生。且LPL基因具有多态性,有研究证明LPL等位基因缺陷的杂合子携带者由于葡萄糖储存受损而降低了全身胰岛素敏感性,这可能与血浆甘油三酯(triglyceride,TG)浓度增加和血浆中不同水平的改变有关[13]。
ApoC3是LPL的天然抑制剂,由于其对LPL和肝脏摄取残余脂蛋白的双重抑制活性,ApoC3已成为调节富含TG的脂蛋白血浆水平的目标靶点之一[14⁃15]。因LPL的表达通过对抗磷脂酶A的作用,从而减少胰岛素分泌;LPL还可以通过其酰基转移活性,促进甘油二酯的生成,进一步调节胰岛素分泌。在ApoC3基因缺陷的小鼠体内,LPL活性增加,更易患肥胖,并形成严重的肝脏IR。ApoC3缺乏可导致脂肪组织增加TG衍生脂肪酸的摄入量,脂肪组织质量增加,逐渐导致IR。例如,ApoC3缺乏可能影响脂肪组织的各种内分泌因子(如瘦素、电阻素和脂肪素)的分泌,这些激素已知影响胰岛素敏感性,与脂肪组织质量相关[16]。
1.3 ApoC3可抑制胰岛素信号转导通路并激活内质网应激和炎症反应 内质网应激(endoplas⁃mic reticulum stress,ERS)是指在内质网功能的稳态失调时出现错误折叠与未折叠蛋白质在内质网腔内聚集的状态。ERS和细胞内炎症反应的激活被认为是IR的重要细胞生物学机制。对骨骼肌ApoC3过表达小鼠的研究显示,ERS和炎症标志物水平升高最终导致核因子κB激酶抑制剂/核转录因子κΒ途径的激活,通过使丝氨酸残基中的胰岛素受体底物⁃1磷酸化来减弱胰岛素信号传导途径,并增加炎症基因的转录。与此相符,还发现细胞外调节蛋白激酶1/2抑制阻止了由ApoC3引起的ERS和炎症的改变,以及胰岛素信号通路的减弱[17]。ApoC3对ERS和炎症反应的激活是通过Toll样受体2(toll⁃like receptor 2,TLR2)介导的,TLR2不仅识别许多含脂质的分子,而且识别内源性蛋白质,它在骨骼肌细胞中表达,并参与脂肪酸诱导的IR。ApoC3在骨骼肌中还可以通过TRL2介导的细胞外调节蛋白激酶1/2激活,抑制过氧化物酶体增殖物激活受体γ⁃1和AMP活化蛋白激酶的活性,从而减弱肌管中胰岛素信号传导并导致ERS的活化。ApoC3还可通过磷酸化丝氨酸残基中的胰岛素受体底物⁃1,抑制丝氨酸/苏氨酸蛋白激酶磷酸化来减弱此胰岛素信号传导途径,并下调脂肪细胞中的胰岛素受体水平[18]。上述研究结果证实了ApoC3在IR发病机制中的作用,并且为IR的治疗提供了可能的新靶点。
1.4 ApoC3可诱导胰腺β细胞凋亡 T2DM的IR与胰岛β细胞衰竭联系之中涉及ApoC3的作用[19]。除肝脏产生的ApoC3水平升高可导致IR外,ApoC3的局部表达升高亦可增强胰岛局部的IR,并促进胰岛β细胞的凋亡。糖尿病患者除了血清ApoC3水平升高外,胰岛局部的IR使得胰岛内的ApoC3水平升高,进一步使得细胞质内Ca2+浓度增加,从而在胰岛β细胞凋亡中发挥了不利作用[19⁃20]。ApoC3可通过SR⁃B1和酪氨酸激酶的激活,使得L型电压门控Ca2+通道过度激活,进而影响胰岛β细胞的凋亡。ApoC3通过增加L型电压门控Ca2+通道的电导率和密度,进而增加细胞质内Ca2+的浓度,从而导致胰岛β细胞凋亡;加入人抗ApoC3抗体后,细胞质Ca2+浓度变化以及其对胰岛β细胞凋亡的影响得到了逆转[17]。此外,ApoC3水平升高可通过激活丝裂原活化蛋白激酶p38和细胞外信号调节蛋白激酶1和2,增加磷酸化水平,促进胰岛β细胞的凋亡[21]。但ApoC3诱导的Ca2+通道上调的确切分子机制还有待进一步研究。
上述研究研究均表明,ApoC3水平的升高,可导致胰岛β细胞的凋亡,并进一步引起IR,并可通过多种信号通路实现,但现无统一观点,值得进一步探讨。
1.5 ApoC3的基因多态性 基因多态性是指在一个生物群体中,同时和经常存在两种或多种不连续的变异型或基因型或等位基因。人类基因多态性在阐明人体对疾病、毒物的易感性与耐受性,疾病临床表现的多样性,以及对药物治疗的反应性上都起着重要的作用。
ApoC3基因位于人第11号染色体长臂q23区,长约311 kb,由4个外显子和3个内含子组成,与载脂蛋白A1、A4基因紧密相连,共同组成一个基因家族。ApoC3的基因多态性与某些疾病的发病风险相关,对于其基因多态性位点,也有较多的报道,但是不同的学者有不同的观点,且所涉及的学科也较为广泛。
ApoC3基因的启动子区包括5个基因多态性位点,ApoC3⁃482C>T基因多态性位于ApoC3启动子区胰岛素抵抗反应元件附件附近,它的多态性变化与部分血浆脂质及载脂蛋白的水平相关,尤其是与IR密切相关。对胰岛素调节的丢失定位于T⁃455C和C⁃482T,这两者间存在相互作用;经转染发现,这两个多态性位点中的任一位点单碱基改变便即可对任何浓度的胰岛素缺乏反应,使得ApoC3的过度表达,导致IR。ApoC3基因启动子区的一个功能区⁃胰岛素反应元件内的两个基因多态性位点rs2854116和rs2854117的突变增加了肝脏对乳糜微粒的吸收进而促成了肝脏脂肪的储积及IR。ApoC3基因中单倍型区块1的A⁃T⁃C⁃C等位基因与1型糖尿病发病率显着增加相关,其风险随着该等位基因拷贝数的增加而增加。在ApoC3启动子中的⁃641c等位基因中,其同源性较高,与ApoC3水平明显降低和胰岛素敏感性增加相关[22]。
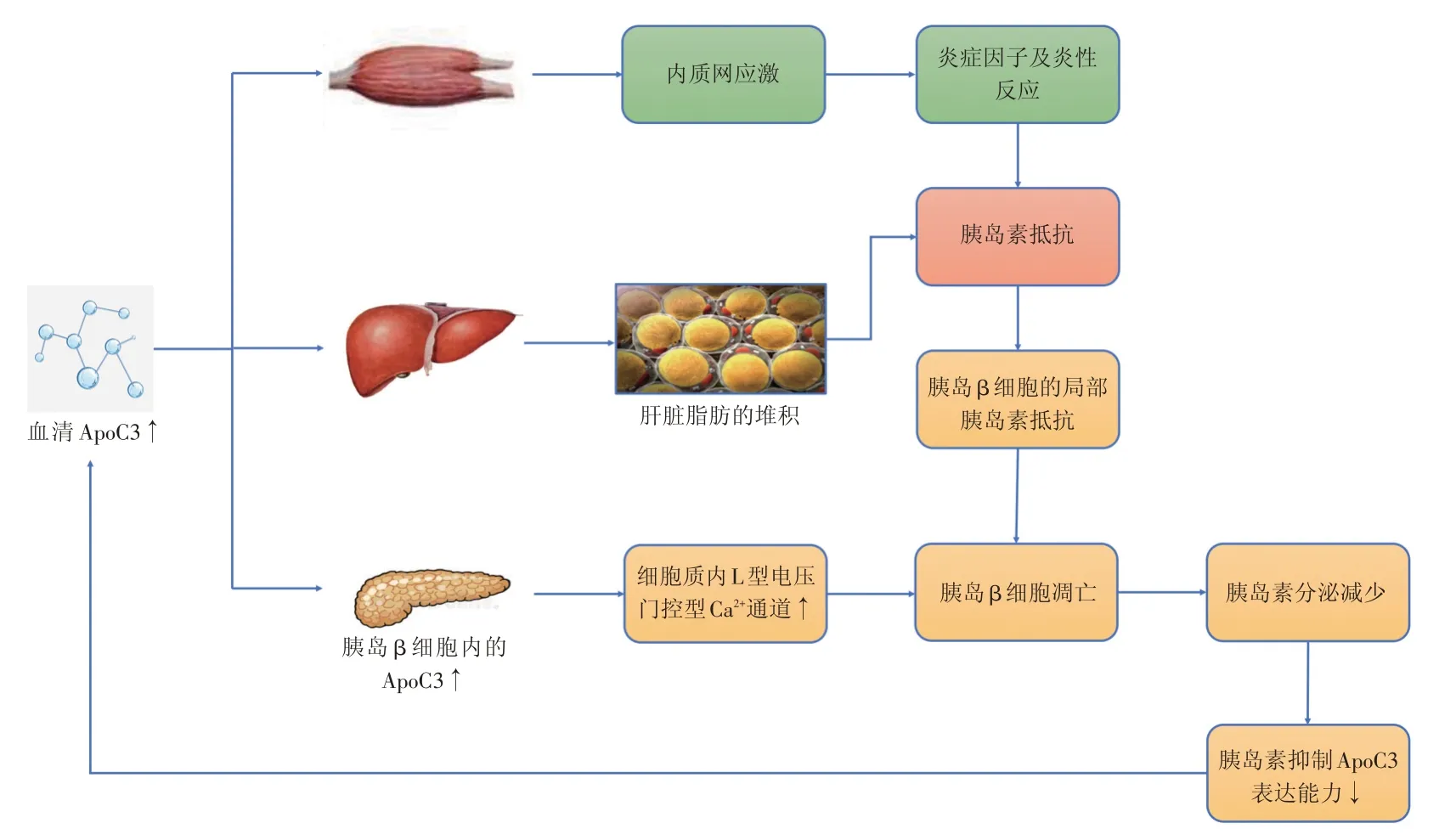
图1 ApoC3与IR相关的机制Fig.1 Mechanism of the relation between ApoC3 and IR
2 ApoC3在IR相关疾病中的研究进展
ApoC3在各种IR相关疾病中的浓度有相似的变化,其升高见于多种疾病情况,如:PCOS、T2DM、AS。
2.1 ApoC3与PCOS PCOS是育龄期女性最常见的内分泌疾病。IR在PCOS妇女中普遍存在,且IR型PCOS患者存在更为严重的糖脂代谢紊乱。有学者提出,PCOS的发生与血清ApoC3水平的升高有关[23⁃24]。李荔等[24]证实ApoC3在PCOS患者的血清中高表达。通过研究,发现多毛组血浆ApoC3较对照组表达水平显著升高,其血清ApoC3表达水平亦显著升高,并推测ApoC3可作为PCOS患者代谢异常的治疗靶点[25]。
通过提取PCOS患者卵巢颗粒细胞研究发现PCOS与PI3K/AKT信号通路有关,而ApoC3是此信号通路的下游靶位点[26]。有学者[27]通过敲除肝细胞中上述信号通路中的叉头转录因子1(forkhead box protein O1,FOXO1),发现与正常的肝细胞相比,其糖代谢和细胞生存都受到影响。FOXO1能够促进糖异生基因葡萄糖-6-磷酸酶和磷酸烯醇式丙酮酸羧激酶的表达,同时可结合到ApoC3的启动子上,促进ApoC3的表达,加重代谢紊乱[28]。人类肝细胞中转染FOXO1后ApoC3的表达升高;ApoC3负向调控FOXO1导致细胞胰岛素敏感性下降,最终导致IR。在循环中,ApoC3抑制了LPL的活性,导致VLDL的TG成分水解和吸收减少,延长了VLDL在循环中的持久性。同时,在IR状态时,FOXO1活性不能被抑制,从而导致血糖和TG升高。因此,ApoC3可通过PI3K/AKT/FOXO1信号通路影响PCOS患者IR的形成与发展,从而导致严重的血糖血脂代谢紊乱,引起远期并发症。
2.2 ApoC3与T2DM IR和胰岛β细胞功能障碍被认为是T2DM发展的关键因素;然而,将这两个缺陷联系起来的分子机制仍不清楚。越来越多的研究发现,T2DM可能与调节失调的ApoC3的合成和分泌有关,血清ApoC3是代谢综合征的独立危险因子。
从人群研究[29]中发现,我国回族及汉族的T2DM患者血清ApoC3水平较正常人群高,且差异有统计学意义,这也证实了血清ApoC3对T2DM发病有促进的作用。同时,在前瞻性研究中,血清ApoC3水平的升高被认为是T2DM的独立预测指标[30⁃32]。从机制上而言,IR的发生与ERS启动胰岛β细胞的凋亡有关,ApoC3参与其中。在T2DM的发展过程中,胰岛素可以由胰岛β细胞分泌。一个新的证据支持在β细胞中线粒体自噬对T2DM的作用,在氧化应激的持续状态下,线粒体自噬作用和ER功能障碍逐渐损害β细胞功能,并促使凋亡途径激活;β细胞质量的下降加剧了β细胞改变对胰岛素分泌的损害[33⁃34],最终导致IR。当体内的胰岛素需求量超过胰岛β细胞的分泌能力时,内质网中未折叠蛋白的增加产生ERS,通过ApoC3介导TRL2激活细胞外调节蛋白激酶1/21/2,影响胰岛素信号通路传导,导致胰岛β细胞的凋亡[17,35⁃36]。
此外,线粒体功能与胰岛素敏感性之间存在相关性。在患有IR或T2DM者中以及在该疾病的小鼠模型中观察到骨骼肌和白色脂肪组织中的线粒体基因表达和氧化能力受损,提出线粒体功能降低可能是IR发展的潜在原因。线粒体氧化脂质的能力降低,将会导致脂质中间体如二酰基甘油和神经酰胺在细胞内积累,并通过激活新型蛋白激酶C来抑制胰岛素信号传导[37]。线粒体损伤也会损害白色脂肪细胞的内分泌和脂肪生成的功能,并促成全身IR的发展。同时,研究表明内质网与线粒体之间存在接触位点,称为线粒体相关的ER膜,以维持细胞稳态。此膜允许线粒体和ER之间交换代谢物和离子,因此每个分子都受到另一个分子的氧化状态的影响。ApoC3可通过B族1型清道夫受体和酪氨酸激酶的激活,使得L型电压门控Ca2+通道过度激活。ER内腔充当Ca2+存储的位点这是线粒体中活性氧信号的介体[38]。在ERS细胞中,Ca2+从ER释放,并被线粒体吸收,从而间接增加线粒体中活性氧。在线粒体和ER之间氧化还原平衡的调节,Ca2+起着至关重要的作用[39⁃41]。
T2DM的特征在于IR,胰岛素释放主要是由高血糖刺激的。当β细胞经常暴露于高血糖症时,葡萄糖代谢会增强,线粒体电子传输链衍生的线粒体中活性氧增加,并诱导、加重ERS。线粒体中产生的线粒体中活性氧诱导ERS,促进线粒体中活性氧生成,导致恶性循环,并在β细胞中建立了氧化应激状态,β细胞无法充分应对高血糖症。线粒体中活性氧破坏细胞成分,包括脂质,蛋白质和脱氧核糖核酸,并触发促进IR的转录变化。氧化应激改变分子和细胞成分会影响细胞机制,例如自噬或炎症。这些过程被改变或破坏,这导致氧化应激和IR的进一步增加。
血清中ApoC3水平的升高,可以抑制下丘脑中LPL的表达,进而影响糖脂代谢,导致肥胖和IR。中枢神经系统可表达数种脂肪酶,以在局部水解TG。LPL是TG的水解酶,并在大鼠海马体和下丘脑内高度表达,并通过增强大脑脂质来调节小鼠代谢稳态的重要调节剂[42]。LPL促进下丘脑中脂肪酸的吸收,从而代谢脂肪酸,产生脂肪酸衍生的长链脂肪酸CoA的积累。在小鼠脑室内注射ApoC3可以强烈抑制下丘脑LPL活性[43]。增强的LPL活性可能激活下丘脑脂质敏感途径,通过增加长链的神经元摄取脂肪酸,调节能量平衡和体重;ApoC3能拮抗这些作用,表明ApoC3诱导肥胖的可能机制和碳水化合物体内平衡的恶化有关。但需更多证据来确定ApoC3在中枢神经系统上的丰度和活性。
综上,ApoC3可通过参与线粒体的氧化应激、ERS以及调节LPL表达过程,调节机体稳态,促使T2DM患者IR的发生,并进一步影响预后。
2.3 ApoC3与AS AS是由于血管内皮脂质代谢紊乱和内皮损伤,继而引发血管壁的不同程度病变的一类慢性进行性病理生理过程。有学者[3]通过对20例冠状动脉无狭窄的心脏病患者和195例冠状动脉有狭窄的心脏病患者的血清ApoC3水平的比较分析发现,血清ApoC3浓度在冠脉狭窄患者中高表达;并推测ApoC3可以作为心血管疾病的潜在独立风险因子。
血管内皮是胰岛素作用的靶组织之一,胰岛素与血管内皮细胞胰岛素受体结合后,通过胰岛素受体底物⁃1和PI3K/AKT信号通路激活内皮型一氧化氮合酶(endothelial nitric oxide synthase,eNOS),eNOS合成NO调节血管的正常生理功能。有学者[44⁃45]通过动物实验发现,ApoC3可促进AS形成,其机制与整体氧化应激水平升高及动脉壁氧化应激和ERS水平增加有关。在内皮型一氧化氮合酶eNOS信号通路中,ApoC3可抑制胰岛素活性,抑制血管内皮中NO的产生,而NO有舒张血管和抗AS作用。胰岛素可活化eNOS,刺激产生NO,而血管内皮的IR状态使其功能失调。有研究发现[46⁃47],ApoC3能在内皮细胞内抑制胰岛素的信号传导而发生IR,同时抑制NO释放,使血管内皮处于紧张状态、难以舒张,导致血管内皮细胞损伤。内皮细胞受损、活化、募集白细胞进而发生粘附,这是AS发生的初始环节。血管内皮细胞黏附分子的诱导产生以及对循环单核细胞的募集促使AS产生和斑块不稳定性,富含ApoC3的脂蛋白是心脑血管疾病的重要危险因素,ApoC3不仅抑制LPL的脂解活性,而且激活血管内皮细胞,产生血管细胞黏附分子,促使单核细胞黏附和转移,构成AS的早期炎症反应。ApoC3诱导血管内皮细胞表达血管细胞黏附分子⁃1和细胞间黏附分子⁃1,募集循环中的单核细胞并发生黏附,可通过蛋白激酶Cβ和NF⁃κB激活血管内皮细胞。
因此,ApoC3是一个独立的且具有促进炎症和AS发生发展作用的蛋白,不仅可通过调节脂代谢,而且可通过其直接致炎及致AS作用影响动脉粥样硬化性心血管疾病的发展。
3 总结与展望
IR的发生机制错综复杂,涉及多因素的相互作用、相互影响,但其确切机制尚未阐明。种种研究及其现象表明血清ApoC3的升高可导致IR的发生发展密切相关。已有研究表明,ApoC3在脂蛋白代谢中起着关键作用,但它也会通过多种机制影响碳水化合物的代谢,从而导致糖代谢异常,其机制包括胰岛β细胞凋亡、IR增强及高血糖的发展等。在分子水平上,ApoC3的基因具有多态性,亦有研究表明相关位点突变与IR及糖脂代谢异常密切相关。ApoC3水平升高可通过激活丝裂原活化蛋白激酶p38和细胞外信号调节蛋白激酶1和2,使得Ca2+通道密度增多,促进胰岛β细胞的凋亡,促进IR发生。且FOXO1是ApoC3的重要启动子特定DNA区域,其表达的增强与ApoC3水平的增加有重要作用;但是其相关具体信号通路未明,更多的推测可能与胰岛素信号传导障碍有关,其中PI3K/AKT通路是最多研究者探讨的通路。同时,氧化应激和ERS也是ApoC3导致IR发生的新兴热点,也使得ApoC3成为治疗IR的重要治疗靶点之一,是治疗IR的一个突破点,值得我们进一步探讨和挖掘。
