新型漆酚基异羟肟酸衍生物的合成及HDAC抑制活性研究
2020-03-09齐志文陈虹霞王成章
周 昊,齐志文,陶 冉,陈虹霞,王成章*
(1.中国林业科学研究院 林产化学工业研究所;生物质化学利用国家工程实验室;国家林业和草原局林产化学工程重点实验室;江苏省生物质能源与材料重点实验室,江苏 南京 210042;2.南京林业大学 江苏省林业资源高效加工利用协同创新中心,江苏 南京 210037)
组蛋白去乙酰化酶(HDAC)是国内外公认的治疗癌症的重要靶点。HDAC异常导致的组蛋白乙酰化状态失衡与肿瘤的发生和发展有密切关系,已经在大多数肿瘤细胞中发现HDAC的过度表达。HDAC抑制剂通过改变细胞内乙酰化水平,从而在调节基因的翻译过程、抑制细胞生长、诱发细胞凋亡或分化、抑制肿瘤细胞血管再生等方面产生抗肿瘤活性[1]。设计开发低毒、高效的HDAC抑制剂类似物已成为国内外抗肿瘤靶向药物研究的热点。按照结构特点,HDAC抑制剂可分为4大类:异羟肟酸类、苯甲酰胺类、亲电酮类和环肽类[2]。其中,异羟肟酸类化合物具有特异性强、抗肿瘤活性好且毒副作用小等优势,是目前研究最广泛且最深入的一类HDAC抑制剂,典型的异羟肟酸类HDAC抑制剂结构按功能可分为3部分:表面识别区、连接臂区和锌离子结合区。表面识别区主要由疏水片段构成,通常是苯环衍生物;连接臂区为脂肪链;锌离子结合区的功能基团为异羟肟酸[3-4]。研究[2]表明:异羟肟酸基团是HDAC抑制剂的关键药效基团,其可直接与HDAC酶的Zn2+结构结合,从而有效抑制HDAC的活性。目前,Merck公司的异羟肟酸类HDAC抑制剂Vorinostat(SAHA)已被美国FDA批准用于治疗淋巴癌,此外,还有多个异羟肟酸类HDAC抑制剂处于临床研究阶段[5-6]。漆酚来源于我国天然产物生漆,漆酚是一种具有邻苯二酚结构的烷基酚类化合物,其侧链为不饱和C15的烷烃,漆酚具有很好的抗肿瘤生物活性,其对人体9种器官29种肿瘤细胞均有抑制作用[7-8],但是漆酚结构不稳定,容易氧化聚合,限制了其作为抗肿瘤药物的开发与应用[9-11]。Ryckewaert等[12]的研究表明不饱和漆酚具有一定的HDAC抑制活性,其结构与FDA批准使用的HDAC抑制剂SAHA结构相似,但是还缺少HDAC抑制关键结构单元锌离子结合区。因此本研究拟通过醚化反应阻断漆酚氧化聚合,通过Diels-Alder、水解和羟氨化等反应,在漆酚侧链尾部引入异羟肟酸基团,并且在其苯环或烷基链引入不同药效基团,合成不同的漆酚基异羟肟酸衍生物,评价其对HDAC 2的抑制活性,采用分子模拟软件对漆酚基异羟肟酸衍生物与HDAC 2进行分子对接研究,分析二者之间的相互作用,为天然产物漆酚用于HDAC抑制剂的开发提供重要理论基础。
1 实 验
1.1 原料、试剂与仪器
漆酚,实验室自制,取一定量生漆原料,以1 ∶20(g ∶mL)甲醇超声波作用10 min后,减压回收溶剂,即得精制漆酚,纯度大于90%。HDAC检测试剂盒购买于上海酶联生物科技有限公司;甲醇(色谱纯),石油醚、乙酸乙酯、二氯甲烷等试剂均为分析纯。IR采用德国TENSOR 27型FT-IR红外光谱仪测定;1HNMR和13CNMR谱采用德国BRUKER 500 MHz核磁共振仪测定(TMS为内标);质谱采用安捷伦LC-QTOF-MS高分辨质谱仪测定。
1.2 漆酚基异羟肟酸衍生物的合成
1.2.1亚甲基醚漆酚异羟肟酸(化合物1) 将3.2 g漆酚溶于50 mL N,N-二甲基甲酰胺(DMF)和50 mL 二氯甲烷(DCM)中,加入0.8 g NaH,氩气保护下回流过夜,冷却至室温后缓慢加入100 mL水,DCM萃取(60 mL ×3),饱和食盐水洗后,无水硫酸钠干燥,减压蒸干,柱层析(石油醚(PE)洗脱)得亚甲基醚漆酚。取1.2 g亚甲基醚漆酚,溶于2 mL甲苯,加入2 mL丙烯酸酯,氩气保护下加热回流36 h,减压蒸干溶剂,加入10 mL乙醇,1.2 g KOH,加热回流10 min,减压蒸干溶剂,加入10 mL水,乙酸乙酯(EA)萃取,无水硫酸钠干燥,减压蒸干,柱层析,洗脱剂V(PE) ∶V(EA)=3 ∶1,得中间产物S1。
将30 mg S1 溶于1 mL无水DMF 中,加入30 μL的N,N-二异丙基乙胺(DIPEA),110 mg 硅基保护羟胺,57 mg 2-(7-氧化苯并三氮唑)-N,N,N′,N′-四甲基脲六氟磷酸盐(HATU),反应搅拌过夜,加压蒸干溶剂,将产物溶于2 mL TBAF-THF 溶液中,室温搅拌40 min,减压蒸干,直接柱层析,洗脱剂V(DCM) ∶V(MeOH)=50 ∶1,得目标产物化合物1。
1.2.28′-羟基亚甲基醚漆酚异羟肟酸(化合物2) 取410 mg化合物S1溶于5 mL 二氯甲烷,加入250 mg 间氯过氧苯甲酸(mCPBA),室温搅拌过夜,饱和碳酸氢钠溶液洗涤(5 mL ×3),无水硫酸钠干燥,减压蒸干得无色油状物,将其全部溶于2 mL 无水四氢呋喃中,缓慢滴加到5 mL 含500 mg 氢化铝锂混悬液中,升温至70 ℃反应2 h,冷却至室温,缓慢加入0.5 mL 水后,再加入0.5 mL 15%氢氧化钠溶液,再次滴加1.5 mL 水,搅拌30 min后过滤,5 mL 四氢呋喃洗涤,滤液减压除去四氢呋喃,加入3 mL 水,乙酸乙酯萃取(3 mL×4),饱和食盐水洗涤,无水硫酸镁干燥,硅胶柱层析,洗脱剂V(PE) ∶V(EA)=2 ∶1,得中间产物S2。
取60 mg S2 溶于0.5 mL 丙酮中,冰水浴降温,滴加入200 μL Jones试剂(三氧化铬-硫酸水溶液),室温反应30 min,加入100 μL异丙醇,搅拌20 min,加入2 mL 饱和氯化钠溶液,EA萃取(2 mL ×5),无水硫酸钠干燥,硅胶柱层析,洗脱剂V(PE) ∶V(EA)=1 ∶1,得中间产物S3。
取55 mg S3 溶于0.5 mL 甲醇中,加入10 mg NaBH4,室温反应30 min,减压蒸干溶解,加入3 mL EA 溶解,饱和食盐水洗涤两次,水洗液用EA 萃取(3 mL ×2),无水硫酸钠干燥,加压蒸干溶剂,真空干燥2 h,加入1 mL DCM,13 mg四氢吡喃基羟胺(NH2OTHP),15 mg 1-乙基-(3-二甲氨基丙基)碳酰二亚胺盐酸盐(EDC·Cl),20 μL三乙醇胺(TEA),反应过夜,5% 柠檬酸水溶液洗涤(2 mL ×2),水相DCM 萃取(3 mL×2),合并有机相,减压蒸干溶剂,加入0.8 mL 甲醇溶解,冰水浴降温,加入1 mL 10% HCl 溶液,反应1 h,减压蒸干溶剂,加入2 mL 甲醇溶解,微孔滤膜过滤,得目标产物化合物2。
1.2.36-硝基亚甲基醚漆酚异羟肟酸(化合物3) 将620 mg化合物S1溶于5 mL新蒸醋酐中,降温至0 ℃,剧烈搅拌下加入300 mg无水硝酸铜,2 h后再加入300 mg无水硝酸铜,再过3 h后加入10 mL水,搅拌20 min后用乙酸乙酯萃取(10 mL×2),有机层用饱和碳酸氢钠溶液洗涤(20 mL×2),无水硫酸镁干燥;减压蒸干得淡黄色油状物,将其溶于10 mL甲醇中,加入0.5 mL水,1.1 g KOH,加热回流3 h后减压蒸干溶剂,再加入5 mL水,乙酸乙酯萃取(5 mL×3),饱和食盐水(10 mL)洗涤,无水硫酸镁干燥,减压蒸干得淡黄色油状物,将油状物45 ℃真空干燥5 h后溶于5 mL无水二氯甲烷中,加入160 mg NH2OTHP,400 mg O-苯并三氮唑-N,N,N′,N′-四甲基脲四氟硼酸(TBTU),0.6 mL N,N-二异丙基乙胺(DIPEA),室温反应6 h,10%柠檬酸水溶液洗涤(5 mL×2),之后饱和碳酸氢钠溶液洗涤(10 mL×2),无水硫酸钠干燥,减压蒸干溶剂;硅胶柱层析(V(PE) ∶V(EA)=2 ∶1)得无色油状物,将其溶于3 mL甲醇中,加入0.5 mL 6 mol/L盐酸溶液,室温搅拌2 h,减压蒸干溶剂,加入3 mL 水,乙酸乙酯萃取(3 mL ×3),饱和碳酸氢钠溶液(5 mL)洗涤,之后饱和食盐水(5 mL)洗涤,无水硫酸钠干燥,减压蒸干溶剂得淡黄色油状物,再将油状物溶于1 mL甲醇中,微孔滤膜过滤,HPLC纯化后,得到目标产物化合物3。
化合物1~3的合成路线见图1。
1.3 分子对接研究
采用GLIDE程序Version10.2将化合物分子与HDAC 2晶体结构(PDB ID:4 LXZ)进行对接;从PDB蛋白质数据库中下载HDAC 2蛋白的晶体结构,对接前对HDAC 2晶体结构进行调整键序、添加氢原子、填充缺失的侧链和环、去除水分子等预处理,并用OPLS_2005 力场对其结构进行能量最小化优化。配体化合物通过LigPrep模块被最小化,产生适当的电离、互变异构、立体化学和环构象,采用MMFFs力场计算部分原子电荷。采用Glide模块生成一个受体栅格文件,该文件利用下载的晶体结构中的原始配体确定活性位点的位置和大小,并模拟化合物配体与HDAC 2的结合模式。Glide对接采用额外精度(XP)模式和默认协议,配体被视为灵活。所有对接化合物均按Glide评分函数进行评分,并进一步分析其结合方式和与关键氨基酸的相互作用。
1.4 HDAC 2酶抑制活性
通过HDAC检测试剂盒AK-501检测化合物1~3对HDAC 2的抑制活性,将样品和阳性对照药伏立诺他(SAHA)用Tris缓冲液(50 mmol/L,pH值8.0)分别配制成质量浓度20、4、0.8、0.16 mg/L的溶液,于96孔板上每孔依次加10 μL缓冲液、 10 μL样品溶液、 5 μL酶溶液和25 μL底物溶液,空白孔加25 μL缓冲液和25 μL底物溶液,对照孔加20 μL缓冲液、 5 μL酶溶液和25 μL底物溶液,加毕,37 ℃下孵化30 min,加入显色剂50 μL,再在37 ℃下孵化15 min,静置后用Thermo(Multiskan Go)酶标仪在405 nm波长条件下读取吸光值,计算各化合物对HDAC 2的抑制率和IC50值。抑制率=(对照孔平均OD值-化合物孔平均OD值)/对照孔平均OD值。
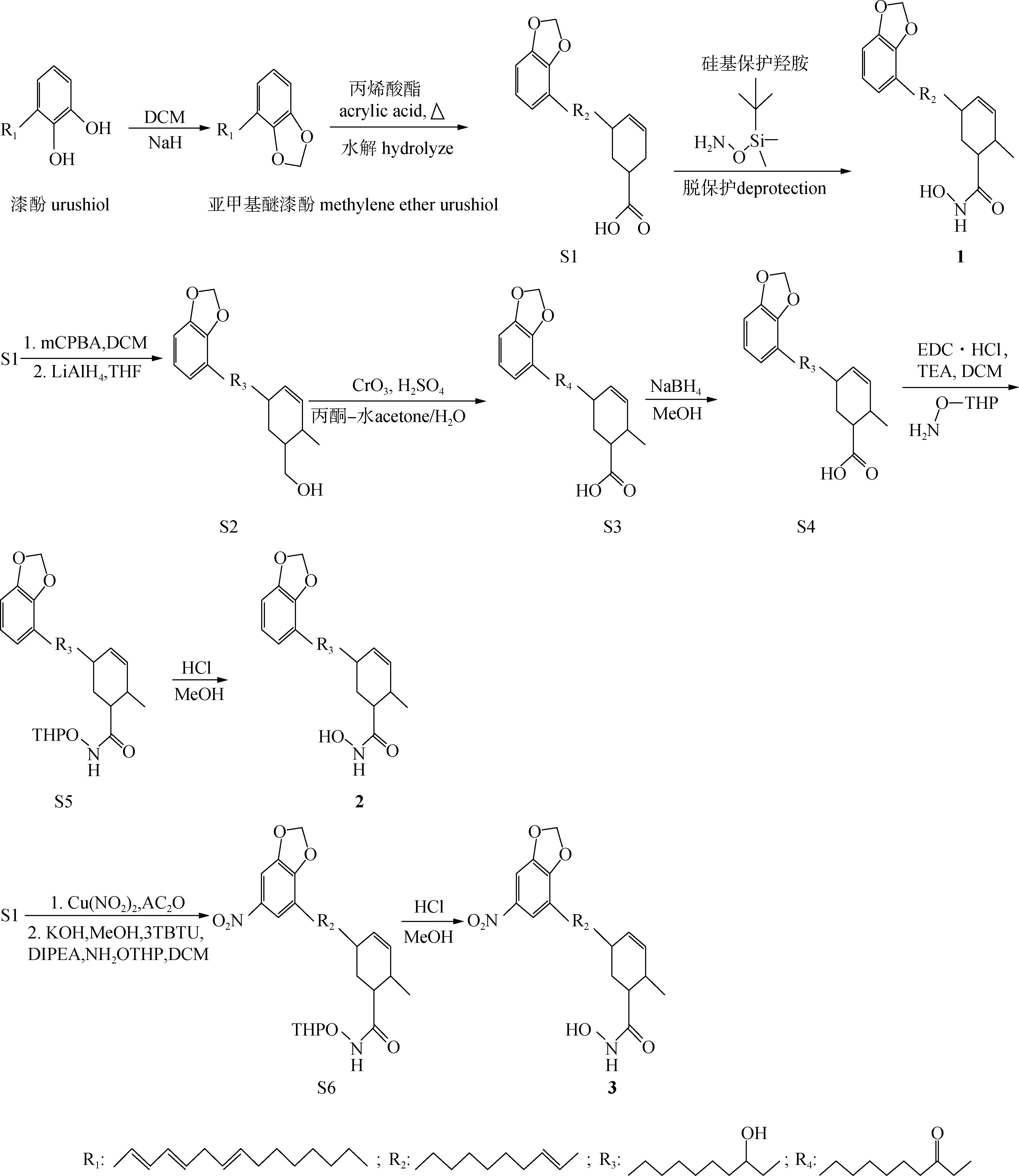
图1 目标化合物1-3的合成路线图
2 结果与讨论
2.1 化合物的合成及结构鉴定
所有目标化合物均未见文献报道,合成的目标化合物结构经1H NMR、13C NMR、 ESI-MS和IR确证。以三烯烷基漆酚为起始原料,与二氯甲烷和NaH进行醚化反应,生成亚甲基醚漆酚,可有效阻断漆酚的氧化聚合。利用漆酚烷基侧链的共轭双键与丙烯酸酯进行Diels-Alder反应,再通过水解得到具有环己烯甲酸结构的关键中间体S1,将S1与O-(叔丁基二甲基硅烷)羟胺和HATU进行反应,即可得到侧链尾部具有异羟肟酸基团的目标化合物1。为了考察漆酚苯环和脂肪链上引入功能基团对HDAC抑制活性的影响,合成了目标化合物2和3。以中间体S1为原料,先与mCPBA反应,使脂肪链上双键氧化成酮,再通过氢化铝锂还原成羟基,但同时S1侧链尾部的羰基也被还原成羟基,因此继续通过与Jones试剂进行氧化反应,与NaBH4进行还原反应得到中间体S4,将S4与NH2OTHP反应,即得到脂肪链上具有羟基结构的目标化合物2。以中间体S1为原料,先与无水硝酸铜进行反应,使苯环上引入硝基,再与NH2OTHP反应,即得到苯环上具有硝基结构的目标化合物3。

2.2 分子对接研究
采用Grid评分函数评价化合物与HDAC 2的对接效果(图2),Glide评分是通过计算蛋白质和配体之间氢键、静电相互作用、范德华力、疏水作用等,对这些作用力进行综合评判得到对接分数,Glide评分是筛选活性和非活性化合物的一种很好的方法[13-14]。
化合物1、2和3的对接分数分别为-7.724、-7.914和-7.946,表明3种化合物均能很好地与HDAC 2的活性口袋结合。由对接结合模式(图3)可以看出,3种化合物的脂肪链部分占据了口袋中长而窄的管道部位,异羟肟酸基团都位于管道的底部,异羟肟酸基团中羟基和羰基可与HDAC 2酶的组氨酸145(His145)和酪氨酸308(Tyr308)形成氢键相互作用,其中羟基都可和口袋底部的Zn2+形成稳定螯合。

图2 FDA批准的异羟肟酸型HDAC抑制剂(SAHA)和三烯烷基漆酚的结构图
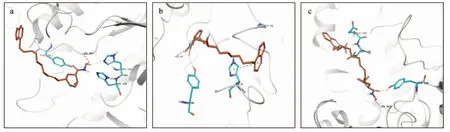
a.化合物1 compound 1;b.化合物2 compound 2;c.化合物3 compound 3
此外化合物3苯环上的硝基还可与谷氨酸103(Glu103)和天冬氨酸 104(Asp104)形成氢键作用。化合物与HDAC结合中的氢键和疏水相互作用,在稳定配合物和提高结合亲和力方面起着至关重要的作用[15]。3种化合物中化合物2和3显示出较高的Glide评分,这表明在漆酚的烷基侧链中引入电子供体基团羟基或在苯环上引入硝基等取代基,可显著提高其对HDAC 2的结合亲和力,因为它们能与残基形成更强的氢键相互作用。
2.3 HDAC2酶抑制活性

图4 不同浓度化合物对HDAC 2抑制率的影响 Fig.4 Effect of different concentrations of compounds on inhibition rate of HDAC 2
不同浓度的3种化合物和阳性药SAHA对HDAC 2的抑制效果如图4所示。可以看出,化合物1、2和3对HDAC 2均起到了良好的抑制作用,抑制率均随着浓度的增加呈上升趋势,呈现出明显的浓度依赖性。
当质量浓度为20 mg/L时,化合物1、2和3对HDAC 2的抑制率为94.2%、96.2%和97.6%,其对HDAC 2的半数抑制质量浓度(IC50)为0.33、0.29和0.24 mg/L。化合物2和3对HDAC 2的抑制效果要优于化合物1,其IC50值和阳性药SAHA(IC50=0.20 mg/L)的相当。3种化合物物对HDAC 2的抑制结果与分子对接结果一致,表明在漆酚的烷基侧链中引入供电子基团羟基,或在苯环上引入硝基等取代基可显著提高其对HDAC 2的抑制活性。
3 结 论
3.1通过醚化反应阻断漆酚氧化聚合,再经Diels-Alder、羟氨化等反应,在漆酚侧链尾部引入异羟肟酸基团,在其苯环或脂肪链引入硝基、羟基等官能团,合成了3种新型亚甲基醚漆酚异羟肟酸衍生物,分别是亚甲基醚漆酚异羟肟酸(化合物1)、8′-羟基亚甲基醚漆酚异羟肟酸(化合物2)和6-硝基亚甲基醚漆酚异羟肟酸(化合物3),并用1H NMR,13C NMR和MS对合成的目标化合物进行了确认。
3.2目标化合物与HDAC2的分子对接结果显示:化合物1、2和3的对接分数分别为-7.724,-7.914和-7.946,均能很好地与HDAC 2的活性口袋结合,异羟肟酸基团中羟基和羰基可与HDAC 2酶的His145和Tyr308形成氢键相互作用,并能与活性口袋底部的Zn2+形成稳定螯合,化合物3苯环上的硝基还可与Glu103和Asp104形成氢键作用。
3.3通过试剂盒AK-501检测化合物对HDAC2的抑制活性,结果表明:化合物1、2和3对HDAC 2均起到了良好的抑制作用,其对HDAC 2的半数抑制质量浓度(IC50)分别为0.33、0.29和0.24 mg/L。其中化合物2和3对HDAC 2的抑制效果要优于化合物1,其IC50值和阳性药SAHA(IC50=0.20 mg/L)的相当。说明在漆酚的脂肪链中引入羟基或在苯环上引入硝基等可提高其对HDAC 2的亲和力和抑制活性,本研究可为漆酚基HDAC抑制剂的开发提供重要的参考。
