叉头框蛋白O1在内毒素血症急性肾损伤中的作用*
2020-03-03张梦希李志莲校振萌李锐钊陈源汉梁馨苓
张梦希, 董 伟, 李志莲, 校振萌, 3, 李锐钊, 陈源汉, 梁馨苓, 3△
[1南方医科大学第二临床医学院, 广东 广州 510515; 2广东省人民医院(广东省医学科学院)肾内科, 广东 广州 510080; 3华南理工大学医学院, 广东 广州 510006]
急性肾损伤(acute kidney injury,AKI)是威胁人类健康的常见危重症,其特征是肾脏损伤及肾功能急剧下降。据统计,在ICU中,AKI的发生率可高达38%~51%[1],其中脓毒血症AKI的发生率约占全部类型AKI的50%,目前已经成为我国社区获得性AKI的首位原因,院内获得性急性肾损伤的第2位原因[2]。脓毒血症AKI预后不良,目前临床尚缺乏防治脓毒血症AKI的有效药物,因此找寻脓毒血症AKI发病机制中的新靶点具有重要的临床意义。内毒素及其所触发的炎症因子瀑布效应对肾小管上皮细胞损伤是引起脓毒血症AKI的重要因素[3],但具体分子机制尚不十分明确。叉头框蛋白O(forkhead box protein O,FOXO)是FOX家族的一个亚群,从蠕虫到人类均有表达。该亚群有4个成员,分别是FOXO1、FOXO3、FOXO4和FOXO6,其中,FOXO1是FOXO家族中最早发现的成员之一[4]。FOXO1作为一种转录因子,可与细胞核内DNA上的反应元件结合,激活靶基因,在DNA损伤/修复、应激、血管生成、糖代谢和肿瘤发生等多过程中发挥着关键性的作用[5]。既往研究发现,FOXO1可在足细胞、肾小管上皮细胞和系膜细胞等多种肾实质细胞中广泛表达,并在糖尿病肾病、肾脏纤维化及狼疮性肾炎等多种疾病中发挥作用[6-8]。但是在内毒素血症AKI中,关于FOXO1的研究尚缺乏,本研究旨在探讨肾小管上皮细胞FOXO1是否参与了内毒素血症AKI的发生及其相关机制。
材 料 和 方 法
1 主要材料及试剂
24只6~8周龄雄性C57BL/6小鼠,购自广州中医药大学实验动物中心(合格证号:No.44005800008550)。人近端肾小管上皮HK-2细胞购自美国模式培养物集存库。DMEM/F12和胎牛血清(Gibco);抗FOXO1抗体、山羊抗兔 II 抗和DAPI(Cell Signaling Technology);抗过氧化物酶体增殖物激活受体γ辅激活因子1α(peroxisome proliferator-activated receptor γ coactivator-1α, PGC-1α)抗体和抗Bax抗体(Abcam);抗GAPDH抗体(Bioworld);脂多糖(lipopolysaccharide,LPS; Sigma,Escherichiacoli055:B5);Ad-FOXO1(汉恒生物);MTT(MP Biome-dicals);MitoTracker Red CMXRos和MitoSOX Red(Invitrogen);血清肌酐(serum creatinine, SCr)试剂盒(Cayman);血尿素氮(blood urea nitrogen, BUN)试剂盒(索莱宝)。
2 方法
2.1内毒素血症AKI小鼠模型的构建 采用随机数表法将24只小鼠随机分为4组(n=6):空白对照(control)组、6 h组、12 h组和24 h组。后3组分别在小鼠腹腔注射LPS(10 mg/kg)后的6 h、12 h和24 h行眼球取血并处死,control组小鼠腹腔注射生理盐水。LPS浓度设置及时点选取参考以往的文献报道[9]。
2.2HK-2细胞的培养 配制含5%胎牛血清的DMEM/F12完全培养基,将细胞置于37 ℃、5% CO2的恒温培养箱中培养,使用0.05%胰酶(含EDTA)进行传代,每4~5 d传代1次。取处于对数生长期并且状态良好的细胞进行后续实验。
2.3FOXO1过表达腺病毒感染实验 将HK-2细胞接种于6孔板,待HK-2细胞融合率达40%~50%,弃去培养基,6孔板每孔加2 mL无血清培养基及1 μL病毒液,培养24 h后换液,改用完全培养基继续培养48 h后收集细胞进行检测。
2.4MTT法检测HK-2细胞活力 取对数生长期的HK-2细胞接种于96孔板,分为对照组及LPS(10、20、40和80 mg/L)组。每组设置5个复孔,同时设置空白孔(在96孔板中直接加入培养基),药物干预结束后,每孔加入20 μL MTT溶液(5 g/L,即0.5% MTT)继续培养4 h,培养结束后小心吸出孔内培养液,每孔加150 μL二甲基亚砜,置摇床上避光振荡10 min,酶标仪检测各孔在560 nm处的吸光度值(A值)。细胞相对活力(%)=(处理组A值-空白孔A值)/(对照组A值-空白孔A值)×100%。
2.5MitoTracker染色 将细胞种植于共聚焦显微镜专用培养皿里,待细胞完全贴壁后进行染色。弃去旧培养基,加少量无血清培养基冲洗3次,每皿加入500 μL MitoTracker Red CMXRos工作液(1∶5 000,200 nmol/L),于37 ℃孵箱中避光孵育25 min,弃去染液,PBS洗3次,最后加入500 μL无血清培养基孵育细胞,准备用激光共聚焦显微镜拍照。正常细胞内线粒体呈丝线状,而损伤时的线粒体可出现肿胀及片段化改变[10]。
2.6Western blot实验 每个样本取50 μg总蛋白,使用8% SDS-PAGE,上样后100 V电泳90 min,PVDF膜200 mA转膜120 min。5%脱脂奶粉室温封闭1 h,TBST洗膜后加入 I 抗[分别为 FOXO1(1∶1 000)、PGC-1α(1∶1 000)、Bax(1∶1 000)和GAPDH(1∶5 000)]4 ℃孵育过夜。TBST 洗膜3次,每次5 min, II 抗(1∶3 000)室温孵育1 h。洗膜后ECL法显色。使用ImageJ软件对Western条带灰度进行半定量分析,以GAPDH为内参照。
2.7RT-qPCR实验 根据TRIzol说明书操作一步法提取总RNA,采用核酸定量仪 NanoDrop Lite测定样本RNA浓度,且A260/A280比值应为1.8~2.0,提示样本中RNA纯度合格。样品-80 ℃保存备用。各引物序列见表1。

表1 RT-qPCR引物序列
逆转录反应体系为20 μL。反应条件为:37 ℃,15 min;85 ℃,5 s,1个循环。PCR扩增程序为:95 ℃预变性5 min;95 ℃变性10 s、60 ℃退火25 s、72 ℃延伸25 s,此3步共循环40次;72 ℃终止5 min。每个样本设2个复孔,定量结果取平均值。
2.8MitoSOXTM染色 用13 μL二甲基亚砜溶解50 μg MitoSOXTM试剂,配制5 mmol/L储存液。使用时用无血清培养基稀释成5 μmol/L工作液,6孔板每孔加1.0~2.0 mL,于37 ℃孵育10 min,PBS洗3次,每次5 min,准备用激光共聚焦显微镜拍摄。
3 统计学处理
用SPSS 20.0统计软件进行分析。数据均采用均数±标准差(mean±SD)表示,多组间比较采用单因素方差分析(one-way ANOVA),组间两两比较采用LSD法。以P<0.05为差异有统计学意义。
结 果
1 内毒素血症AKI时小鼠肾脏FOXO1 mRNA及蛋白表达下调
小鼠腹腔注射LPS(10 mg/kg)后6 h、12 h和24 h行眼球取血并处死,获取小鼠血清及肾组织标本,结果显示,小鼠SCr和BUN随时间延长呈上调趋势,且在24 h上升最为显著(P<0.05),见图1A;小鼠肾组织病理染色结果显示,LPS腹腔内注射24 h后,皮质及外髓质肾小管上皮细胞出现轻微的肿胀及空泡样变性、管腔狭窄,见图1B;肾组织电镜可见,LPS 24 h组小鼠肾小管上皮细胞内线粒体肿胀、嵴消失,空泡样变性,见图1C。上述结果提示LPS可诱导小鼠出现内毒素血症急性肾损伤。Western blot结果显示,与对照组相比,LPS各组FOXO1蛋白表达量下调,且在24 h组下调最为显著(P<0.05),见图1D。FOXO1在肾组织中广泛表达,小鼠肾组织免疫荧光可见FOXO1在肾小管(尤其是近端肾小管)上皮细胞中表达量较高,在肾小球中表达量相对较低。与对照组相比,LPS诱导的内毒素血症AKI小鼠肾组织中FOXO1下调(红色荧光示FOXO1,蓝色示细胞核),见图1E。提取小鼠肾皮质RNA,RT-qPCR结果显示FOXO1转录水平下降(P<0.05),见图2。上述结果提示,肾小管上皮细胞FOXO1可能参与了内毒素血症AKI的发生发展。与此同时,线粒体氧化磷酸化相关基因(PGC-1α、线粒体复合体I亚基Ndufs1和线粒体复合体V亚基Atp5o)的转录均下调(P<0.05),见图2。
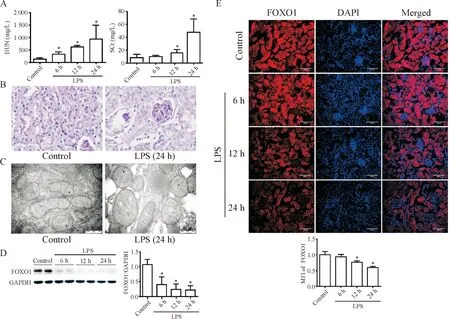
Figure 1. The expression of FOXO1 in kidney of mice was down-regulated during LPS-induced AKI. A: blood urea nitrogen (BUN) and serum creatinine (SCr) were increased after LPS injection; B: periodic acid-Schiff (PAS) stainning (×400) indicated vacuolar deformation and swelling of renal tubular epithelial cells; C: electron microscopy (×39 000) showed that mitochondrial injury in tubular epithelial cells, as exhibited by the disorder or disappearance of crista, and mitochondrial swelling and deformation, was aggravated in the LPS group; D and E: Western blot and immunofluorescence (×400) revealed that FOXO1 protein in mouse kidney tissue was down-regulated over time. MFI: mean fluorescence intensity. Mean±SD.n=6.*P<0.05vscontrol group.
图1 内毒素血症AKI时,小鼠肾脏FOXO1表达下调

Figure 2. The mRNA expression of FOXO1, PGC-1α, Ndufs1 and Atp5o was down-regulated during LPS-induced AKI. Mean±SD.n=6.*P<0.05vscontrol group.
图2 内毒素血症AKI时,小鼠肾脏FOXO1及线粒体氧化磷酸化相关基因mRNA表达下调
2 LPS诱导HK-2细胞FOXO1 mRNA及蛋白表达下调
为检测内毒素血症AKI对肾小管上皮细胞FOXO1表达的影响,本研究用LPS(浓度为40 mg/L)干预HK-2细胞48 h。Western blot实验结果发现LPS可诱导HK-2细胞内FOXO1蛋白的表达量下调(P<0.05),见图3A。RT-qPCR结果提示,FOXO1的mRNA也出现下调(P<0.05),见图3B。同样地,免疫荧光结果显示,与对照组相比,LPS处理组的HK-2细胞内FOXO1荧光强度较弱(P<0.05),见图3C。这些体外实验结果与前述动物实验所观察到的现象一致,因而进一步证明了内毒素血症可诱导肾小管上皮细胞FOXO1 mRNA及蛋白表达水平下降。
3 过表达FOXO1减轻LPS诱导的HK-2细胞线粒体损伤
LPS可诱导HK-2细胞内FOXO1下调,同时引起线粒体生物合成关键调控分子PGC-1α下调以及促凋亡因子Bax上调;利用过表达腺病毒载体对人FOXO1基因进行构建及包装,并用该病毒感染HK-2细胞,结果发现FOXO1过表达上调了PGC-1α,并下调了Bax蛋白水平(P<0.05),见图4A。采用不同浓度(10、20、40和80 mg/L)的LPS刺激HK-2细胞时,细胞活力呈不同程度下降,而FOXO1过表达腺病毒组在不同LPS浓度条件下的细胞活力均高于空病毒载体组(P<0.05),见图4B。这提示FOXO1过表达可逆转LPS诱导的肾小管上皮细胞活力下降。利用活细胞线粒体染料Mitotracker标记HK-2细胞线粒体,发现LPS刺激可引起肾小管上皮细胞线粒体呈现片段化改变,而过表达FOXO1可减轻LPS引起的这种线粒体片段化(红色示线粒体),见图4C。MitoSOXTM染色可标记线粒体超氧化物,反映线粒体氧化呼吸功能损伤程度。LPS刺激下,HK-2细胞线粒体产生大量超氧化物,MitoSOXTM染色呈强阳性;FOXO1过表达后,超氧化物产生量较LPS刺激时减少,见图4D。此外,RT-qPCR分析发现,LPS刺激HK-2细胞可引起线粒体复合体I亚基Ndufb8和线粒体复合体V亚基Atp5g1这两个线粒体氧化磷酸化相关基因的mRNA水平下调,而过表达FOXO1可减轻该下调程度(P<0.05),见图4E。由此可见FOXO1过表达可减轻LPS引起的HK-2细胞线粒体损伤及氧化磷酸化功能障碍。

Figure 3. FOXO1 protein and mRNA expression was down-regulated in the HK-2 cells induced by LPS. Western blot (A), RT-qPCR (B) and immunofluorescence (C; ×400) for deteting the expression of FOXO1 in HK-2 cells that have undergone different interventions. MFI: mean fluorescence intensity. Mean±SD.n=3.*P<0.05vscontrol group.
图3 LPS诱导HK-2细胞内FOXO1转录及表达下调
讨 论
LPS是革兰氏阴性菌内毒素,是脓毒血症炎症反应的主要致病因子之一。LPS不仅可触发大量炎症因子瀑布效应,而且可通过活化Toll样受体4(Toll-like receptor 4,TLR4)直接诱导肾小管上皮细胞损伤[11],因此采用LPS构建内毒素血症模型可以有效且相对合理地模拟脓毒血症时内毒素对机体的损伤作用。然而,内毒素血症引起AKI的具体分子机制尚不明确。既往研究发现,FOXO家族在肾脏中广泛表达,参与多种肾脏疾病的发生发展。在糖尿病肾病中存在FOXO1表达及活化障碍并参与疾病发生及进展,足细胞过表达FOXO1可通过活化PTEN诱导激酶(PTEN-induced kinase,PINK)/parkin通路减轻高糖诱导的线粒体自噬和足细胞凋亡[6];此外,过表达FOXO1还可减轻高糖诱导的系膜细胞PGC-1α下调及线粒体损伤[12]。同时,肾小管上皮细胞过表达FOXO1还可抑制小管间质纤维化(tubulointerstitial fibrosis,TIF)相关分子如信号传导及转录激活蛋白1(signal transducer and activator of transcription 1,STAT1)的表达,从而减轻TIF及小管细胞凋亡[8]。但尚无FOXO1在内毒素血症AKI中的研究。本研究首次发现,内毒素血症AKI时肾小管上皮细胞FOXO1 mRNA及蛋白表达下调;并且在细胞模型中,过表达FOXO1可以逆转LPS诱导的肾小管上皮细胞活力下降,减轻线粒体损伤和氧化磷酸化功能障碍,因此我们推测肾小管上皮细胞FOXO1表达下调可能是内毒素血症AKI的发病机制之一。
肾脏是人体第二大能量需求器官,其线粒体含量仅次于心脏。在肾小管上皮细胞中线粒体含量十分丰富。线粒体通过其内膜电子传递链将质子从线粒体基质泵入膜间隙,在内膜上产生电化学梯度,从而驱动二磷酸腺苷(adenosine diphosphate,ADP)磷酸化为三磷酸腺苷(adenosine triphosphate,ATP),用来维持肾小管上皮细胞的电化学梯度及协助溶质运输,以实现肾脏重吸收等重要生理功能[13]。活性氧簇(reactive oxygen species,ROS)是体内一类氧的单电子还原产物,是电子在未能传递到末端氧化酶之前漏出呼吸链并消耗大约2%的氧生成的,当线粒体损伤时,电子传递链功能出现异常,可引起线粒体大量超氧化物产生,对细胞造成氧化损伤[14]。既往研究发现,线粒体损伤是内毒素血症AKI的重要病理生理改变,由此引起的能量代谢异常以及线粒体超氧化物、细胞色素酶C、凋亡激活因子等释放所致的肾小管上皮细胞程序性死亡是重症AKI的关键环节[15]。我们的研究发现,内毒素血症可诱导肾小管上皮细胞活力下降及线粒体损伤,过表达FOXO1可逆转LPS诱导的HK-2细胞活力下降,减轻线粒体损伤,说明FOXO1确实参与了内毒素血症AKI肾小管上皮细胞损伤的发生。那么,FOXO1是通过什么机制参与内毒素血症AKI的呢?FOXO1作为一种重要的转录因子,其下游多种转录产物超氧化物歧化酶2(superoxide dismutase,SOD2)、过氧化氢酶(catalase,CAT)和PGC-1α等均可以影响线粒体能量代谢及再生。其中,PGC-1α是维持线粒体形态及生物合成的“关键调控因子”[16]。文献证实,在急性肾损伤中PGC-1α转录及表达下调,继而引起线粒体功能紊乱及损伤从而介导多种类型AKI的发生[17-18]。在肝细胞等已证实,FOXO1是PGC-1α的上游转录因子[19],并且在糖尿病肾病模型中,也发现过表达FOXO1可以上调PGC-1α[8],但在肾小管上皮细胞中尚不明确FOXO1是否可调控PGC-1α。在我们的研究中,FOXO1过表达可减轻内毒素所诱导的肾小管上皮细胞活性氧增加,同时可增加PGC-1α转录以及PGC-1α下游控制氧化磷酸化的分子——Ndufb8和Atp5g1的转录。因此我们推测,在内毒素血症AKI时肾小管上皮细胞内FOXO1下调可能引起PGC-1α等线粒体功能维持关键因子的转录下调,导致肾小管上皮细胞线粒体损伤及功能障碍,从而介导内毒素血症AKI的发生。具体机制我们需要进一步的研究来验证。
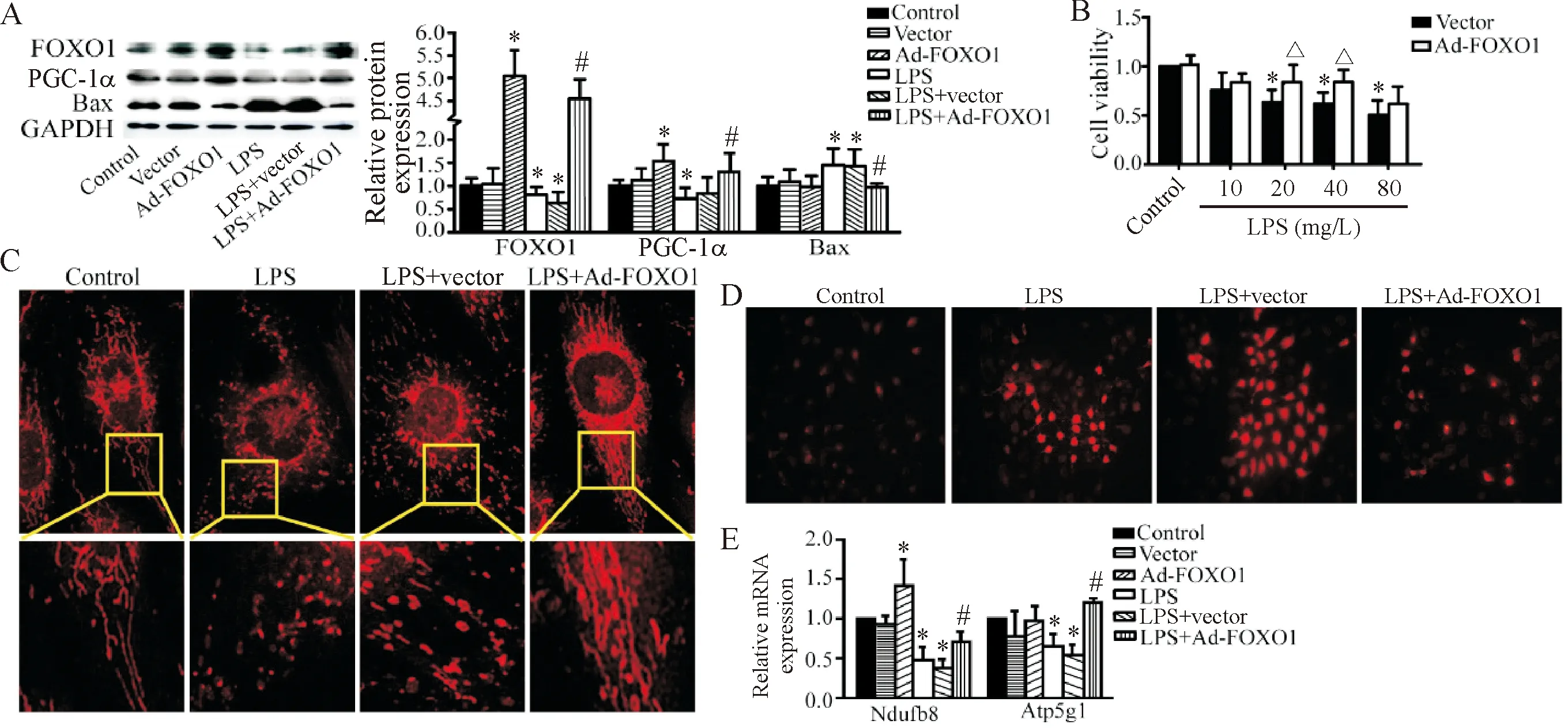
Figure 4. Over-expression of FOXO1 attenuated HK-2 cell injury induced by LPS. A: over-expression of FOXO1 reversed LPS-induced PGC-1α down-regulation and Bax up-regulation in HK-2 cells; B: MTT assay showed that Ad-FOXO1 attenuated LPS-induced cell viability depression; C: MitoTracker staining showed the morphological changes of mitochondria in HK-2 cells (×400); D: MitoSOX staining showed mitochondrial superoxide production in HK-2 cells (×400); E: the mRNA levels of mitochondrial complex I (Ndufb8) and mitochondrial complex V (Atp5g1) were quantified in the mitochondrial fractions. Mean±SD.n=3.*P<0.05vscontrol group;#P<0.05vsLPS group;△P<0.05vsvector group.
图4 过表达FOXO1可减轻LPS诱导的HK-2细胞损伤
综上所述,本研究发现肾小管上皮细胞FOXO1水平下调介导内毒素血症AKI肾小管上皮细胞损伤这一新的机制。该研究结果提供了内毒素血症AKI的新的干预靶点,为内毒素血症AKI的防治提供了新策略。
