核磁共振法测定食品中水苏糖的含量
2019-11-06吉鑫樊双喜杨彤晖刘一诺钟其顶
吉鑫,樊双喜,杨彤晖,刘一诺,钟其顶
1(中国食品发酵工业研究院有限公司,北京,100015) 2 (全国食品发酵标准化中心,北京,100015)
核磁共振(nuclear magnetic resonance,NMR)作为一种有机化合物结构解析的代表性分析方法,在1963年首次应用于定量分析,确定了26种纯有机物质中的分子内质子比率[1]。近年来,NMR定量技术已经在药物分析[2]、代谢组学[3-4]、食品分析鉴别[5]等领域得到了较为广泛的运用。与气相色谱法、高效液相色谱法等传统分析方法相比,NMR仅需要很少的样品量,便可直接测定啤酒[6-7]、果汁[8-9]、葡萄酒[10]和婴儿配方奶粉[11]等不同食品基质中的多种化合物,而无需预先将复杂混合物中的待测物质分离,具有同时测定样品中多种成分含量等优点。
水苏糖是由两个α-半乳糖,一个葡萄糖和一个果糖构成的四糖,为非还原性功能性低聚糖[12]。具有促进肠道内双歧杆菌繁殖及生长、调节肠道内菌群平衡、增强机体免疫力、降血糖和降血脂等功能[13]。由于水苏糖的物化性质较为稳定,且具有良好的生理功能,通常被添加到如饮料、米粉、乳制品、口服液等[14-16]食品中,以适应不同的消费需求。
目前食品中水苏糖含量的检测主要使用行业标准方法(QB/T 4260—2011),即高效液相色谱法[17],另外,采用层析法[18]、气相色谱法[19]等在特定条件下也能检测水苏糖含量。薄层层析法分析时间长、展开剂体积需求大、分离结果差,实验结果准确性、稳定性以及精密度较差;气相色谱法需将糖通过衍生化的方法转换成为可挥发的成分后再测定,过程较为繁琐;高效液相色谱法是较为常用的方法[20],但其分析时间较长,示差折光检测器的稳定性、重复性和选择性均有不足。此外,现行QB/T 4260—2011规定的方法仅适用于以食用植物为原料,经加工提取、精制而成的水苏糖含量进行检测,即P60、P70和P80型水苏糖,对于复杂食品基质中水苏糖的定量分析较为困难,因此,开发一种能够对食品中水苏糖实现快速、准确的定量方法,对于规范水苏糖类产品的市场尤为重要。
基于上述问题,本研究通过引入核磁共振波谱仪,旨在建立基于外标和内标的NMR定量分析方法,实现对食品中水苏糖含量快速、准确地检测。由于食品基质相对复杂,为避免食品中其他物质对内标物质信号峰产生干扰,故选取不同内标物质进行分析,探索NMR法直接应用于食品基质中水苏糖含量定量检测的可行性,弥补现行QB/T 4260—2011高效液相色谱法适用范围不足的问题。
1 材料与方法
1.1 材料与试剂
水苏糖标准品(98%),北京百灵威科技有限公司;叠氮化钠(高纯,NaN3),Biotopped;3-(三甲基硅基)氘代丙酸钠(Sodium-3-(trimethylsilyl)propionate-2,2,3,3-d4,TSP,98%),CIL;KH2PO4,98%,CIL;重水(D2O,99.9%),青岛腾龙微波科技有限公司;柠檬酸(99%),Vetec;烟酰胺(99%),上海安谱实验科技股份有限公司;琥珀酸(99%),北京百灵威科技有限公司;市售含水苏糖的主要食品(样品编号为1-8,其中1号为以草石蚕为原料提取的P70型水苏糖;2号为片剂;3-5号为3种不同品牌的固体饮料,电商平台;6-8号为口服液,超市)。
1.2 仪器与设备
Bruker Avance III HD 400 MHz波谱仪,德国Bruker Biospin公司;Bruker自动进样器(SampleJet),德国Bruker Biospin公司;Bruker SampleJet 5 mm高通量核磁管,德国Bruker Biospin公司;涡旋混合器(MX-S),大龙兴创实验仪器(北京)有限公司。
1.3 方法
1.3.1 缓冲溶液的配制
分别准确称取1.00 g TSP和0.13 g NaN3于烧杯中,用D2O将其溶解并定容于10 mL容量瓶中,得到100 g/L的TSP溶液和13 g/L的NaN3溶液。准确称取8.00 g KH2PO4于烧杯中,用100 mL D2O将其溶解并转移至200 mL容量瓶中,向其中加入5 mL H3PO4、2 mL上述TSP溶液和2 mL NaN3溶液,以及50 mL D2O。静置24 h后,测定该溶液pH值,若pH>2.0,则加入适量H3PO4调整,若pH<2.0,则加入适量KH2PO4固体粉末调整,直至pH稳定到2.0后,完成缓冲溶液的配制。
1.3.2 标准储备液及内标、外标溶液的配制
准确称取0.064 0 g水苏糖标准品于烧杯中,加入适量蒸馏水,待其完全溶解后,转移至10 mL容量瓶中并用蒸馏水定容,摇匀后得到质量浓度为6.4 g/L的水苏糖标准溶储备液。
准确称取适量柠檬酸标准品,配制成2.0 g/L的柠檬酸标准溶液。用移液枪准确吸取100 μL缓冲溶液加入样品管后,取900 μL柠檬酸标准溶液加入样品管并混匀,移取600 μL上述溶液于核磁管中,用于外标法测定。
准确称取适量琥珀酸、烟酰胺标准品,分别配制成10.0 g/L的琥珀酸标准溶液和10.0 g/L的烟酰胺标准溶液,用于内标法测定。
1.3.3 样品制备
准确称取适量1~5号样品于烧杯中,用蒸馏水溶解并转移至容量瓶定容。取2 mL定容后的溶液,使用0.22 μm滤膜过滤。吸取6~8号样品并用蒸馏水稀释至适当浓度。
用移液枪准确吸取100 μL缓冲溶液于样品管中,加入900 μL上述1~8号水苏糖样品溶液,混合均匀后吸取600 μL于5 mm NMR管中,用于外标法测定。用移液枪准确分别吸取100 μL内标溶液和100 μL缓冲溶液于样品管中,加入900 μL上述1~8号水苏糖样品溶液,混合均匀后吸取600 μL于5 mm NMR管中,用于内标法测定。
1.3.41H NMR采样参数
实验所采用的1H NMR的共振频率为400.13 MHz;检测温度300 K(±0.1);空扫次数(DS)为4次;扫描次数(NS)为64次;谱宽(SW)为20.5524;采样点数(TD)为65536;接收增益(RG)为16;弛豫延迟(D1)为4 s。以TSP(δ= 0)作为化学位移的零点。标准的脉冲序列(NOESYGPPR)用于水(δ= 4.8)的信号抑制,有效地减弱了水峰对检测的干扰。
1.3.5 标准曲线的绘制
准确吸取一定体积的水苏糖标准储备液,用蒸馏水逐级稀释,配制成质量浓度分别为6.400、3.200、1.600、0.800、0.400、0.200、0.100、0.050、0.025 g/L的水苏糖标准溶液,并取少量蒸馏水作为空白对照。进行与样品溶液相同前处理后,在1.3.4所述条件下,首先测定柠檬酸外标溶液,再依次对以上9个水苏糖标准溶液进行测定。
1.3.6 精密度分析
本文选取1号样品溶液,1 d内连续测定5次,取平均值,分析方法的日内精密度;连续测定5 d,每天测定5次并取平均值,分析方法的日间精密度。
1.3.7 回收率实验
平行称取25份1号样品,用蒸馏水溶解并转移至容量瓶定容。其中5份作为对照组,其余20份由低至高,分别加入4个不同浓度水平的水苏糖标准品溶液,每个水平重复5次,测定其1H NMR谱图,计算回收率和相对标准偏差。
1.3.8 方法对比
将核磁共振定量法、高效液相色谱行业标准方法QB/T 4260—2011和样品标签标注的水苏糖的含量方法进行对比分析,验证分析方法的准确性。
2 结果与分析
2.1 内标法定量
对于确定的原子,其核磁共振信号强度(峰面积)与产生该信号的原子的数目成正比,而与其化学性质和所处的化学环境无关。以内标物的相对峰面积作为参考,根据样品指定质子共振峰的相对峰面积计算含量,根据以下公式(1)计算分析物浓度[10]:
(1)
式中:ρ,分析物质量浓度,mg/L;M为相对分子质量;nH为质子数;A为信号积分面积;CF为校正因子。
2.2 外标法定量
脉冲宽度定量(pulse length based concentration determination,PULCON)[21]外标法是基于信号强度互易原理,即给定线圈中样品化合物的NMR信号强度与90°脉冲宽度成反比。通过介电特性(离子强度)在样品间的变化来补偿因线圈灵敏度损失的信号强度,将外部参考样品的校准转移到实际样品中。每次定量前,需测定具有已知浓度的用于校准的外部参考样品(如2 g/L柠檬酸),计算定量因子。样品定量公式(2)如下[10]:
(2)
式中:ρ,分析物质量浓度,mg/L;A为绝对积分面积;M为相对分子质量;nH为质子数;NS为扫描次数;P1为1H 90°脉冲宽度;T为检测温度;CF为校正因子。
2.3 校正因子(CF)
校正因子是在NMR波谱仪和测量条件等因素相互影响情况下对样品定量结果的校正,如校正测量过程中对水信号压制的影响等[10]。
校正因子的计算:水苏糖标准品质量与溶液体积之比为纵坐标,外标法或内标法所得的浓度为横坐标,得线性回归y=ɑx+β,ɑ即校正因子(CF)。
2.4 水苏糖1H NMR谱中水苏糖特征峰确认、定量峰以及内标、外标物的选择
以D2O为溶剂,测定水苏糖标准品的1H NMR谱,并对水苏糖1H NMR谱信号峰进行归属。1H NMR(D2O,400 MHz,TSP)δ:5.44(1H,d,J=3.8 Hz),5.00(2H,dd,J=3.5,2.0 Hz),与文献报道一致[24-26]。
由于水苏糖是由1分子α-葡萄糖、1分子β-果糖和2分子α-半乳糖构成的非还原性四糖[24],糖环上多数质子处于相似的化学环境中,如图1-a所示,以D2O为溶剂的水苏糖1H NMR谱中,化学位移分布在δH3.4~4.3范围内的质子信号峰多发生重叠现象,裂分情况难以归属,因此不能用作定量峰进行分析。由于糖端基质子所受的去屏蔽作用明显强于环上其他质子,糖端基质子的化学位移向低场区移动,根据水苏糖结构(图2),2个α-D-半乳糖基的端基质子信号峰(δH5.00)和α-D-葡萄糖基的端基质子信号峰(δH5.44)(图1-b)与样品中其他成分信号峰无重叠,易于辨认。考虑到食品基质的复杂性,故选择葡萄糖基的端基质子信号峰(δH5.44)和α-D-半乳糖基的端基质子信号(δH5.00)作为样品中水苏糖的定量峰,从而确保定量方法的准确性。

a-水苏糖1H NMR谱图;b-水苏糖特征峰图1 以D2O为溶剂的水苏糖1H NMR谱图(a)以及特征峰(b)Fig.1 1H NMR spectra of stachyose in D2O solvent(a) and characteristic peaks(b)

图2 水苏糖结构式Fig.2 Structural formula of stachyose
PULCON外标法定量使用外部参考样品,将外部参考样品的校准转移到实际样品中,因此无需考虑分析物和参考样品之间的相互作用和信号重叠,具有较高的精确度和准确度[27]。此外,采用PULCON外标法成本相对较低,可多次重复利用,进一步降低了核磁共振的检测成本,简化了前处理操作。柠檬酸结构简单,根据1H NMR谱显示,其仅在δH2.65和δH2.52处存在两组双峰,且两组双峰对称,易于辨认,用于定量分析结果准确,因此选择柠檬酸作为外部参考物。
内标法定量需在样品溶液中直接加入一定量的内部标准物。烟酰胺化学性质稳定,在水中具有良好的溶解性,1H NMR谱在δH9.17显示一组双峰,不与水苏糖特征峰重叠且不易与食品基质中其他化合物信号峰发生重叠。琥珀酸结构简单,可溶于水,两个亚甲基所包含的4个质子均处于相同的化学环境中,因此,其1H NMR谱仅在δH2.67处存在单峰,与水苏糖信号峰不发生重叠,峰型尖锐,易于辨认。故选择烟酰胺和琥珀酸作为内部参考物。
2.5 实际水苏糖样品1H NMR图谱
按照1.3.4的条件测定得到水苏糖食品的1H NMR图谱(见图3),上、中、下分别代表的是1号P70型水苏糖样品、4号固体饮料和8号口服液,由图3可知,δH5.44特征信号峰与周围质子信号峰未发生重叠现象。
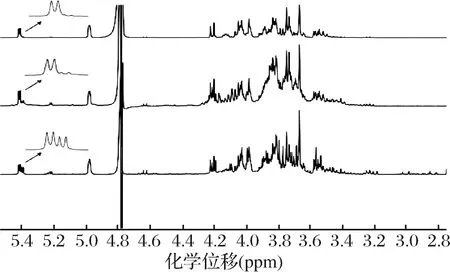
图3 水苏糖样品1H NMR特征峰Fig.3 1H NMR spectra of stachyose sample with characteristic peaks
2.6 方法学考察
2.6.1 线性关系
以水苏糖标准品质量与溶液体积之比为纵坐标,测定浓度为横坐标做线性回归。水苏糖标准品在0.025~6.40 g/L质量浓度范围内线性良好。
柠檬酸作为定量外标,水苏糖回归方程分别为y=1.169 2x-0.0106,R2=1;琥珀酸和烟酰胺分别作为定量内标,水苏糖回归方程分别为y=1.064 3x+0.001 7,R2=0.999 6和y=1.036 5x-0.016 3,R2=1。表明本方法线性关系良好。
2.6.2 精密度分析
选取1号样品溶液,1 d内连续测定5次,每次重复测定5遍,取平均值,分析方法的日内精密度;连续测定5 d,每天测定5次取平均值,分析方法的日间精密度。结果如表1所示,采用PULCON柠檬酸作为外标方法日内精密度和日间精密度分别为1.65%和5.27%,且日内与日间两者水苏糖测定结果无显著性差异(P>0.05),满足测定方法对于精密度的要求。以琥珀酸和烟酰胺作为内部参考物时,方法的日内精密度和日间精密度,结果如表2所示。以琥珀酸作为内标,方法的日内精密度和日间精密度分别为1.34%和0.65%;以烟酰胺作为内标,方法的日内精密度和日间精密度分别为0.83%和2.46%。

表1 柠檬酸作为外标方法日间与日内精密度(n=5)Table 1 Intra-day and inter-day precision of the method with citric acid as an external standard
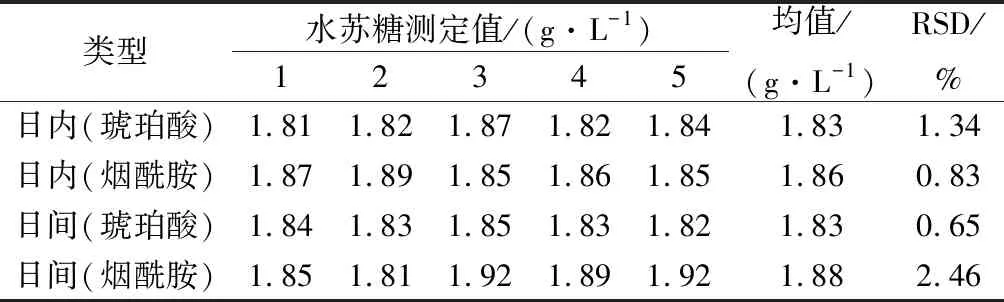
表2 内标方法日间与日内精密度(n=5)Table 2 Intra-day and inter-day precision of internal standard method
2.6.3 回收率实验
选取1号样品溶液,向其中分别添加0.78、1.59、2.34、3.20 g/L,4个不同水平的水苏糖标准溶液,单个水平5次重复取均值,结果取平均值,如表3所示。以柠檬酸作为外标,采用PULCON方法测定4个不同的加标水平下,该方法的回收率在95.63%~101.28%,其平均回收率为97.91%,回收率的RSD在2.00%~2.32%,结果表明该水苏糖检测方法精确度较好,能够满足水苏糖的准确测定。分别以琥珀酸和烟酰胺作为内标对水苏糖进行定量,结果如表4、表5所示。以琥珀酸作为内标测定4个不同的加标水平下,该方法的回收率在96.56%~110.26%,其平均回收率为101.39%,回收率的RSD在1.54%~1.84%。以烟酰胺作为内标测定4个不同的加标水平下,方法的回收率在100.31%~114.10%,其平均回收率为106.35%,回收率的RSD在1.28%~2.22%。

表3 柠檬酸作为外标方法回收率Table 3 Recovery of the method with citric acid as an external standard
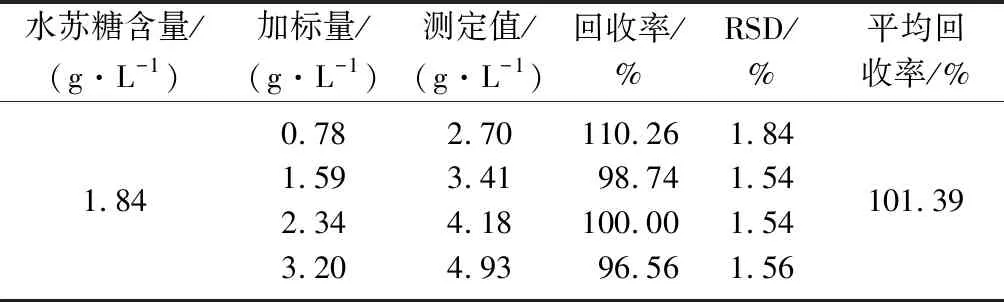
表4 琥珀酸作为内标方法回收率Table 4 Recovery of the method with succinic acid as an internal standard
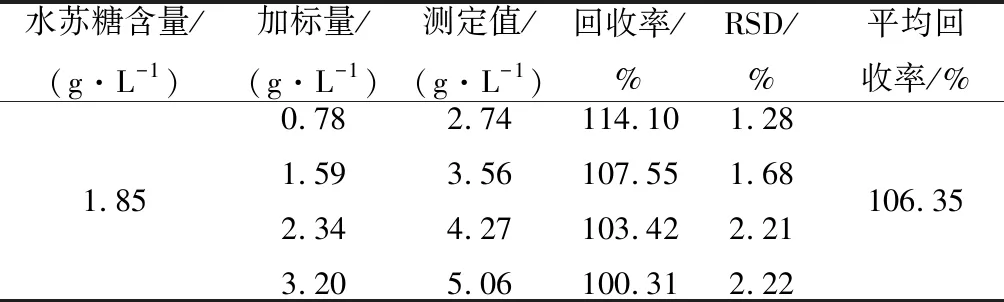
表5 烟酰胺作为内标方法回收率Table 5 Recovery of the method with niacinamide as an internal standard
2.6.4 方法对比
应用上述已经建立NMR定量方法对市售8个样品的水苏糖含量进行定量测定,每个样品采用1H NMR和2D J-resolved进行检测,重复测定2次,取平均值,结果如表6所示。NMR内标和外标定量结果无显著性差异(P>0.05),NMR定量结果符合样品标签注明的含量;2D J-resolved谱结果显示,δH5.44水苏糖信号峰周围不存在可对其定量结果产生干扰的杂质信号,不影响NMR准确定量,结果表明NMR能够用于水苏糖样品的快速定量检测。
采用行标QB/T 4260—2011高效液相色谱法重复测定2次,取平均值,结果如表6所示。1号P70型水苏糖、2号片剂、3号固体饮料和8号口服液样品NMR和高效液相色谱法检测结果基本一致,无显著性差异(P>0.05),进一步表明NMR能够准确定量食品中水苏糖含量;4、5号样品经NMR定量所得结果明显高于行标法和标签注明含量,产生偏差原因可能是样品中其他糖类与水苏糖中葡萄糖基端基质子(δH5.44)信号峰完全重叠;6、7号样品经行标方法检测,未检出水苏糖成分,产生偏差原因考虑主要是样品基质中不含有水苏糖成分而是其他糖类在水苏糖中葡萄糖基端基质子(δH5.44)信号峰处产生信号。
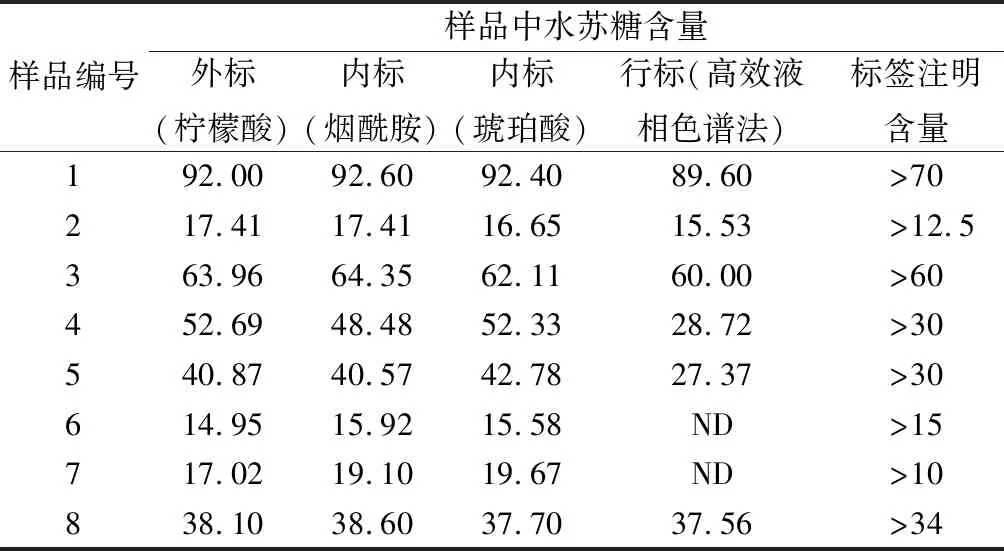
表6 核磁共振和行标高效液相色谱法定量水苏糖结果 单位:%
注:%指单位物质所含水苏糖质量的百分比;ND表示未检出。下同。
为近一步探究4~7号样品NMR定量结果与行标方法产生差异的原因,以水苏糖的半乳糖端基质子(δH5.00)信号作为定量峰对样品中水苏糖含量进行计算。结果如表7所示。

表7 以半乳糖端基质子信号作为定量峰的水苏糖核磁共振定量结果 单位:%
1~3号和8号样品定量结果与以葡萄糖端基质子作为定量峰时结果一致,无显著性差异(P>0.05)。4、5号样品定量结果更接近于行标法测定的水苏糖含量。而6、7号样品在δH5.00处未出现质子特征信号,则认为6、7号样品中不含水苏糖。
根据4、5号样品配料表所注明成分,对其分别添加葡萄糖、果糖、半乳糖和低聚果糖进行加标实验,发现添加低聚果糖的4、5号样品,其δH5.44质子信号积分面积显著增加,由此造成以δH5.44质子信号作为定量峰时,水苏糖定量结果偏高,故δH5.44定量峰不适用于含低聚果糖的食品基质。
3 结论
本研究以柠檬酸作为定量外标,琥珀酸和烟酰胺分别作为定量内标,建立了食品中水苏糖NMR定量分析检测方法。以葡萄糖端基质子(δH5.44)信号作为定量峰,该方法在0.025~6.40 g/L范围内线性良好,相关系数大于0.999;以柠檬酸作为外标方法加标回收率为97.91%,日内精密度与日间精密度分别为1.65%与5.27%;琥珀酸和烟酰胺内标方法加标回收率分别为101.39%和106.35%,日内精密度分别为1.34%和0.83%,日间精密度分别为0.65%与2.46%;对于基质中含有低聚果糖的食品,采用半乳糖端基质子(δH5.00)信号作为定量峰对水苏糖含量进行计算。该方法具有很好的重复性和稳定性。与现行行业标准方法相比,NMR法前处理操作简便,实验过程中避免了有机试剂的引入,单个样品的检测时间为7 min,极大地缩短了待测样品的前处理时间,检测效率显著提高,对食品中水苏糖的定量具有很好的准确性和更广泛的适用性,为含水苏糖食品的市场规范提供了方法支撑。
