巴斯德毕赤酵母鼠李糖代谢基因LRA3功能及其启动子顺式作用元件的研究
2019-10-14梁明丽
梁明丽, 刘 波, 张 伟
中国农业科学院生物技术研究所, 北京 100081
巴斯德毕赤酵母(Pichiapastoris)是甲醇营养型酵母,由于其具有遗传操作简单、胞外分泌表达、高密度发酵及特有的翻译后修饰等优点而成为常用的真核表达宿主[1]。启动子作为表达系统必需且重要的元件显著影响着毕赤酵母表达系统的外源蛋白表达水平[2]。基于启动子PGAP的毕赤酵母表达系统组成型调控外源基因表达,细胞快速生长即表达目的蛋白,使得细胞生长状态和生物量相对诱导型启动子较差,进而降低目的蛋白表达量;此外,当所表达的目的蛋白对宿主细胞生长有损害时,细胞生长状态和生物量更低,无法高效表达目的蛋白。目前,基于甲醇诱导的甲醇氧化酶基因启动子PAOX1的毕赤酵母表达系统得以广泛应用,然而在大规模发酵过程中需要使用易燃的甲醇,存在极大的安全隐患[3];而且,甲醇为有毒物质,不能满足食品及生物医药等安全性要求极高的重组蛋白生产。因此,挖掘以无毒物质为诱导剂的强启动子是完善毕赤酵母表达系统的有效途径。
L-鼠李糖(L-6-脱氧甘露糖)是一种天然六碳糖,是植物果胶和细菌细胞壁的常见组成成分,许多微生物可以利用其作为碳源和能源[4]。目前发现L-鼠李糖代谢主要通过2种途径:磷酸化途径和非磷酸化途径。磷酸化途径存在于如Escherichiacoli、Clostridiumacetobutylicum、Rhizobiumleguminosarum等大多数细菌中;而非磷酸化途径则存在于真菌和少数细菌中,完整的代谢途径基因仅在酵母Scheffersomycesstipitis和Debaryomyceshansenii中阐明[5]。S.stipitis通过4种相关代谢基因(RHA1、LRA2、LRA3、LRA4)编码的酶将鼠李糖依次转化为L-鼠李素-Y-内酯、L-鼠李糖酸、3,6-双脱氧-L-赤式古洛糖,最后转化为丙酮酸和L-乳醛,二者进入中枢代谢途径。
研究发现毕赤酵母在以鼠李糖为唯一碳源的培养基上可以正常生长[6],本实验室前期预测了毕赤酵母鼠李糖代谢途径相关基因,挖掘了2个鼠李糖诱导型启动子PLRA3和PLRA4,其中PLRA3在以鼠李糖为碳源时,转录强度可达PGAP的80%,是基于鼠李糖的强诱导型启动子[6]。由于PLRA3是以无毒鼠李糖为诱导物的强启动子,因此有望替代PAOX1和PGAP应用于工业生产。为进一步提高基于PLRA3的毕赤酵母表达系统的表达效果,需要对表达宿主和表达载体进行优化。本实验室在前期的研究中通过菌株改造工程获得了一株鼠李糖代谢流降低的菌株,在此菌株中PLRA3调控目的蛋白表达量提高了1~2倍[7]。在表达载体优化方面,可以通过PLRA3改良工作提高其转录强度进而增强目的蛋白表达效果。此外,已有研究发现,未诱导状态下,PLRA3表达的毒性蛋白对宿主生长有一定程度的抑制作用,故PLRA3在无鼠李糖诱导时存在微弱的渗漏表达[8]。综上所述,需要对PLRA3进行理性改造以提高PLRA3诱导转录水平、降低渗漏表达,进而提高目的蛋白表达量。基于此,本研究通过基因敲除和回补实验探究LRA3基因与毕赤酵母鼠李糖代谢的相关性;另一方面,本实验室前期研究工作初步确定了PLRA3全长约210 bp,但转录元件未能精确鉴定,本研究应用简单易行的CRRT-PCR方法及生物信息学分析鉴定并预测PLRA3转录相关元件,以期为PLRA3下一步理性改造提供科学指导。
1 材料与方法
1.1 实验材料
1.1.1菌株与质粒 巴斯德毕赤酵母菌株P.pastorisGS115为本实验室保存;pPICZαA质粒购自美国Invitrogen公司;pEASY-Blunt载体及大肠杆菌E.coliTrans1-T1感受态细胞购自北京全式金生物技术有限公司。
1.1.2试剂与仪器 FastPfu DNA聚合酶购自北京全式金生物技术有限公司;琼脂糖购自莫纳生物科技有限公司;琼脂糖凝胶DNA回收试剂盒及质粒小提试剂盒购自天根生化科技(北京)有限公司;8 kb标准Marker(DM8000)购自北京汇百惠生物科技中心;Trizol试剂和Zeocin抗生素购自美国Invitrogen公司;限制型内切酶(AscⅠ、PacⅠ及SwaⅠ)和DNaseⅠ购自美国Thermo scientific公司;RNA 5′焦磷酸水解酶(RNA pyrophosphohydrolase,RppH)、10×Thermopol Buffer、T4 RNA ligase 1及RNase抑制剂均购自美国NEB公司;5×All-In-One RT MasterMix购自加拿大ABM公司;其他试剂均为进口或国产分析纯。
PCR仪器及电穿孔完全系统(美国Bio-Rad公司);立式冷冻离心机(日本HITACHI公司);电泳仪(北京君意东方电泳设备有限公司);凝胶成像分析系统(上海培清科技有限公司);恒温金属浴(金银杏生物科技(北京)有限公司);恒温培养箱(上海一恒科技有限公司);恒温培养振荡器(上海智城分析仪器制造有限公司)。
1.1.3培养基 YPR培养基:2%鼠李糖,2%蛋白胨,1%酵母膏。MD固体培养基:2%葡萄糖,2%琼脂糖,1.34%酵母无氨基氮源(yeast nitrogen base without amino acids,YNB),4×10-5%生物素。MDH固体培养基:2%葡萄糖,2%琼脂糖,1.34% YNB,4×10-5%生物素,4×10-3%组氨酸。MRH固体培养基:2%鼠李糖,2%琼脂糖,1.34% YNB,4×10-5%生物素,4×10-3%组氨酸。
1.2 实验方法
1.2.1LRA3基因的敲除菌株构建 以GS115基因组为模板,利用引物KO0075-FF/KO0075-FR和KO0075-RF/KO0075-RR(引物序列见表1,由生工生物工程(上海)股份有限公司合成)分别PCR扩增LRA3基因的上、下游约500 bp片段作为同源重组臂。PCR体系A如下:GS115基因组 1 μL,5×TansStart FastPfu buffer 10 μL,2.5 mmol/L dNTP 6 μL,FastPfu 1μL,10 μmol/L上、下游引物各2.5 μL,以ddH2O补至50 μL。PCR程序A为:94℃预变性5 min;94℃变性30 s,58℃退火30 s,72℃延伸20 s,共28个循环;72℃终延伸10 min。PCR产物经1%琼脂糖凝胶电泳分析鉴定后,用琼脂糖凝胶回收试剂盒纯化回收。
以pPICZαA质粒为模板,用引物zeocin-F/zeocin-R(表1)扩增zeocin抗性基因表达盒。PCR体系及程序同程序A。

表1 本研究所用引物序列Table 1 Primer sequences used in the study.
以LRA3基因上、下游同源臂及zeocin表达盒片段为模板,利用引物对KO0075-FF/KO0075-RR(表1)进行重叠延伸PCR连接3个片段,PCR体系B如下:LRA3上、下游同源臂及zeocin表达盒片段各1 μL,5×TansStart FastPfu buffer 10 μL,2.5 mmol/L dNTP 6 μL,FastPfu 1 μL,10 μmol/L上、下游引物各2.5 μL,以ddH2O 补至50 μL。PCR扩增条件B为:94℃预变性5 min;94℃变性30 s,52℃退火30 s,72℃延伸75 s,共4个循环;94℃变性30 s,56℃退火30 s,72℃延伸75 s,共25个循环;72℃终延伸10 min。PCR产物经1%琼脂糖凝胶电泳分析鉴定正确后,加入0.8倍体积的异丙醇及0.2倍的3 mol/L醋酸钠,将以上体系混匀于-20℃静止0.5 h后,4℃、12 000 r/min离心10 min,轻轻去除上清后加入同体积75%乙醇清洗2次,晾干后加入15 μL ddH2O,取适量回收产物用1%琼脂糖凝胶电泳分析鉴定。
毕赤酵母GS115感受态细胞制备参考相关文献[9]。将10 μg线性化DNA加入80 μL感受态细胞轻轻混匀,放入预冷电击杯中电击转化,电击完成后将转化体系涂布于含100 μg/mL Zeocin的YPD平板,28℃培养36 h。
提取转化子基因组进行PCR验证,2对验证引物为KO0075-VF/zeocin-R(400)及zeocin-F(400)/KO0075-VR(表1),PCR体系及程序同程序A,PCR延伸时间为1 min。
将PCR验证阳性转化子及野生型GS115划线于MRH平板,观察其生长情况,将LRA3基因敲除酵母菌株命名为GS115-ΔLRA3。
1.2.2LRA3基因的回补 构建LRA3基因回补的同源重组载体pUCSH-LRA3。通过引物HB-F/HB-R(表1)扩增LRA3基因上、下游基因之间片段,PCR体系及程序同程序A,PCR延伸时间为1 min。AscⅠ、PacⅠ双酶切pUCSH载体及PCR产物,利用T4 DNA连接酶连接并转化大肠杆菌克隆菌株Trans1-T1感受态,以HB-F/HB-R为引物进行PCR验证,PCR体系及程序同程序A,PCR延伸时间为1 min。PCR验证正确后送生工生物工程(上海)股份有限公司测序,选择测序正确的质粒进行回补。
质粒经SwaⅠ内切酶线性化后异丙醇醇沉回收。制作GS115-ΔLRA3菌株感受态细胞,将10 μg线性化DNA加入80 μL感受态细胞轻轻混匀,放入预冷电击杯中电击转化,电击完成后涂布于MD培养基,28℃培养36 h。
提取转化子基因组进行PCR验证,通过HB-F/HB-R(表1)验证,PCR体系及程序同程序A,PCR延伸时间为1 min。将PCR验证为阳性的转化子及GS115划线于MRH平板,观察其生长情况。将LRA3基因回补菌株命名为GS115-LRA3。
1.2.3GS115、GS115-ΔLRA3及GS115-LRA3菌株生长情况对比 将GS115、GS115-ΔLRA3及GS115-LRA3划线于MDH、MRH平板,观察3种菌株在以葡萄糖或鼠李糖为唯一碳源的培养基中的生长情况差异。
1.2.4RNA提取、去帽、环化和反转录 挑取GS115单菌落接种于20 mL YPD培养基中,于28℃培养36 h;以1%接种量转接至100 mL YPR培养基中,于28℃培养36 h,离心收集菌体。
GS115总RNA的提取:采用Trizol法抽提毕赤酵母总RNA。用RNase-free的DNase I消化样品中的DNA,以通用的5′-AOX和3′-AOX为引物验证基因组DNA完全去除,PCR体系及程序同程序A,PCR延伸时间为1 min。
RNA去帽:使用RNA 5′焦磷酸水解酶去掉总RNA的帽子结构。反应体系为:10×Thermopol Buffer 5 μL,RNA 8 μL,RppH 5 μL,以RNase-free ddH2O补至50 μL。上下颠倒混匀(或者轻轻吹吸混匀)后,37℃反应2 h,65℃ 5 min终止反应,异丙醇醇沉回收RNA。
RNA环化:使用T4 RNA ligase 1进行RNA环化。反应体系为:10×buffer 2 μL, RNA 500 ng,T4 RNA ligase1 2 μL,RNase抑制剂 1 μL,50% PEG 8000 4 μL,10 mmol/L ATP 3 μL,以RNase-free ddH2O补至20 μL。上下颠倒混匀(或者轻轻吹吸混匀)后16℃温育12 h,65℃ 5 min终止反应。
RNA反转录:用5×All-In-One RT MasterMix进行cDNA反转录。反转录反应体系为:环化RNA 50 ng,5×All-In-One RT MasterMix 2 μL,以RNase-free ddH2O补至20 μL。反应体系混匀后,25℃退火10 min,42℃反应1 h,85℃ 5 min终止反应。
1.2.5嵌套PCR及转录起始位点确定 第一轮PCR反应体系如下:cDNA 100 ng,5×TansStart FastPfu buffer 10 μL,2.5 mmol/L dNTP 6 μL,FastPfu 1 μL,10 μmol/L引物ⅠF/ⅠR(表1)各2.5 μL,以ddH2O补至50 μL。PCR扩增条件为:94℃预变性5 min;94℃变性30 s,58℃退火30 s,72℃延伸1 min,共28个循环;72℃终延伸10 min。
第二轮PCR反应以第一轮PCR产物为模板,引物为ⅡF/ⅡR(表1),PCR体系及程序同第一轮PCR。
将第二轮PCR产物连接至pEASY-Blunt载体后转化E.coliTrans1-T1。以通用引物M13F/M13R进行菌液PCR,PCR体系及程序同嵌套PCR。经1%琼脂糖凝胶电泳分析鉴定选取较长PCR产物所对应的克隆送至生工生物工程(上海)股份有限公司测序。测序结果使用Vector NIT软件分析,确定转录起始位点。
1.2.6生物信息学分析PLRA3利用启动子在线分析工具:Promoter Scan(https://www-bimas.cit.nih.gov/molbio/proscan/)、CpGFinder(http://linux1.softberry.com/berry.phtml?group=programs&subgroup=promoter&topic=cpgfinder)、EMBOSS Cpgplot(https://www.ebi.ac.uk/Tools/seqstats/emboss_cpgplot/)和Nsite (http://linux1.softberry.com/berry.phtml?topic=nsite&group=programs&subgroup=promoter)分析预测PLRA3的转录起始位点、CpG岛及可能的转录调控序列。
2 结果与分析
2.1 LRA3基因敲除同源片段的扩增及菌株构建
以GS115基因组为模板进行PCR,分别扩增LRA3基因的上、下游作为敲除同源臂,同源臂长度约为500 bp;以pPICZαA质粒为模板扩增zeocin抗性基因表达盒,片段大小约为1.2 kb。通过重叠延伸PCR将三者连接构建LRA3基因敲除片段(图1)。

图1 LRA3基因敲除相关DNA片段Fig.1 DNA fragmentation involved in knocking out LRA3 gene.注:M:8 kb标准Marker;1:敲除片段上游同源臂;2:敲除片段下游同源臂;3:zeocin表达盒;4:LRA3基因敲除片段。
通过电击转化将基因敲除片段导入细胞,毕赤酵母的DNA同源重组机制将LRA3基因替换为zeocin抗性表达盒,经Zeocin抗性筛选后再进行基因组水平鉴定(图2)。敲除菌株通过2套引物进行PCR鉴定,上游引物验证结果见图2A,对应菌株的下游引物验证结果见图2B。阳性菌株的上游引物扩增片段大小约为1.2 kb,下游约为1.6 kb,故4号为敲除菌株。将阳性转化子及野生型GS115划线于MRH平板,发现其几乎不生长,将LRA3基因敲除酵母菌株命名为GS115-ΔLRA3。
2.2 LRA3基因回补载体及菌株构建
以GS115基因组为模板进行PCR,扩增LRA3基因上、下游基因之间片段,AscⅠ、PacⅠ双酶切连接至pUCSH载体构建回补载体,PCR验证阳性克隆大小约为2.2 kb(图3A)。线性化DNA电击转化GS115-ΔLRA3感受态,通过缺乏组氨酸的平板筛选阳性转化子后进行PCR鉴定(图3B)。将阳性转化子及野生型GS115划线于MRH平板,发现转化子恢复生长,将LRA3基因回补酵母菌株命名为GS115-LRA3。
2.3 GS115、GS115-ΔLRA3及GS115-LRA3菌株生长情况
将GS115、GS115-ΔLRA3及GS115-LRA3划线于MDH、MRH平板。当以葡萄糖为碳源时,3种菌株均能正常生长;当以鼠李糖为碳源时,GS115和GS115-LRA3正常生长,而GS115-ΔLRA3未见生长(图4),表明LRA3基因参与毕赤酵母鼠李糖代谢过程。
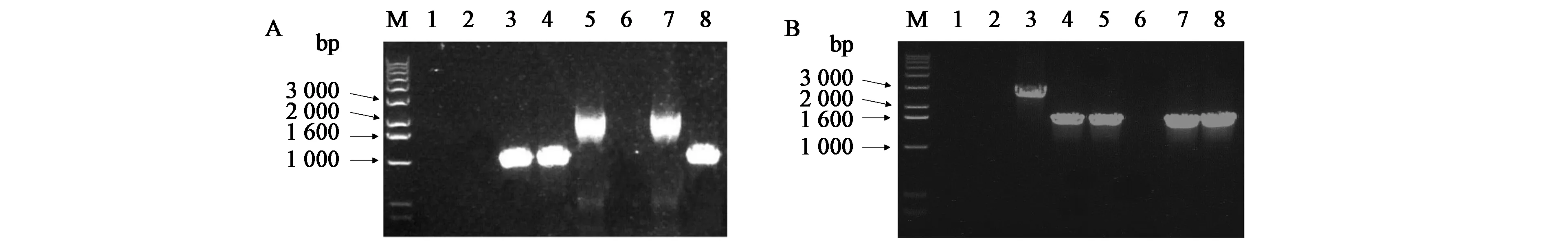
图2 LRA3基因敲除菌株验证Fig.2 Verification of LRA3-disrupted strain.注:A:LRA3基因敲除菌株验证(上游引物);M:8 kb标准Marker;1:GS115基因组;2:阴性对照;3~8:转化子。B:LRA3基因敲除菌株验证(下游引物);M:8 kb标准Marker;1:GS115基因组;2:阴性对照;3~8:对应转化子。
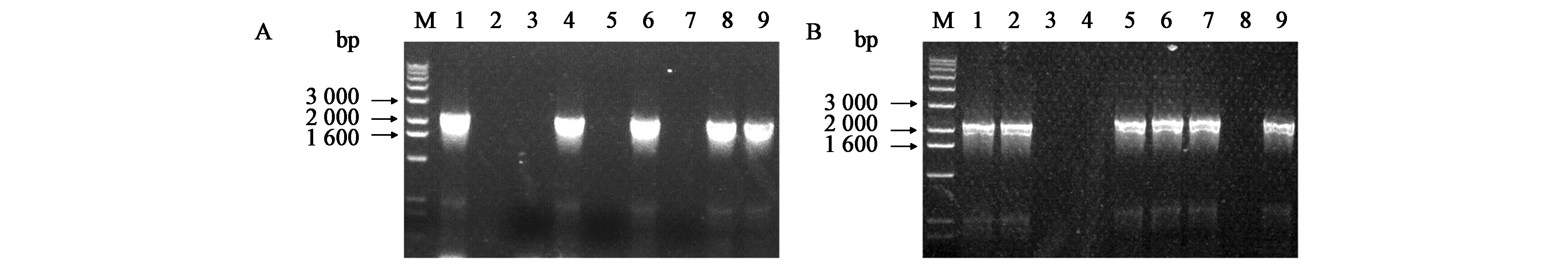
图3 LRA3基因回补载体及菌株验证Fig.3 Verification of the vector for complementing LRA3 gene and LRA3-complemented strain.注:A:LRA3基因回补载体验证;M:8 kb标准Marker;1:GS115基因组;2~9:转化子。B:LRA3基因回补菌株验证;M:8 kb标准Marker;1:回补质粒;2:GS115基因组;3~9:酵母转化子。

图4 3种菌株在不同碳源的生长情况Fig.4 Growth profiles of three strains on different carbon sources.注:左侧培养基碳源为葡萄糖,右侧为鼠李糖;a、b、c分别为GS115、GS115-ΔLRA3、GS115-LRA3。
2.4 GS115总RNA提取及帽子结构去除
通过CRRT-PCR方法确定PLRA3转录起始位点。在以鼠李糖为唯一碳源的培养基中培养GS115,鼠李糖诱导LRA3基因高强度转录。收集菌体,以Trizol法抽提GS115中总RNA。电泳结果显示(图5A),总RNA各条带清晰,未有明显降解。真核生物基因转录时在RNA 5′加帽,促进RNA稳定性。在进行RNA环化前,需要用5′焦磷酸水解酶去除RNA 5′帽子结构(图5B)。
2.5 P. pastoris GS115总RNA环化及嵌套PCR
RNA经RNA环化酶作用后,转录起始位点与RNA 3′多聚腺嘌呤相连而形成环形RNA。根据LRA3的编码区序列,设计2套PCR引物,引物位置示意图见图6A。以环化RNA为模板、以外侧引物(ⅠF/ⅠR)进行PCR扩增,PCR产物理论大小约为1 kb。电泳结果显示,有大小约为1 kb的PCR产物出现(图6B)。
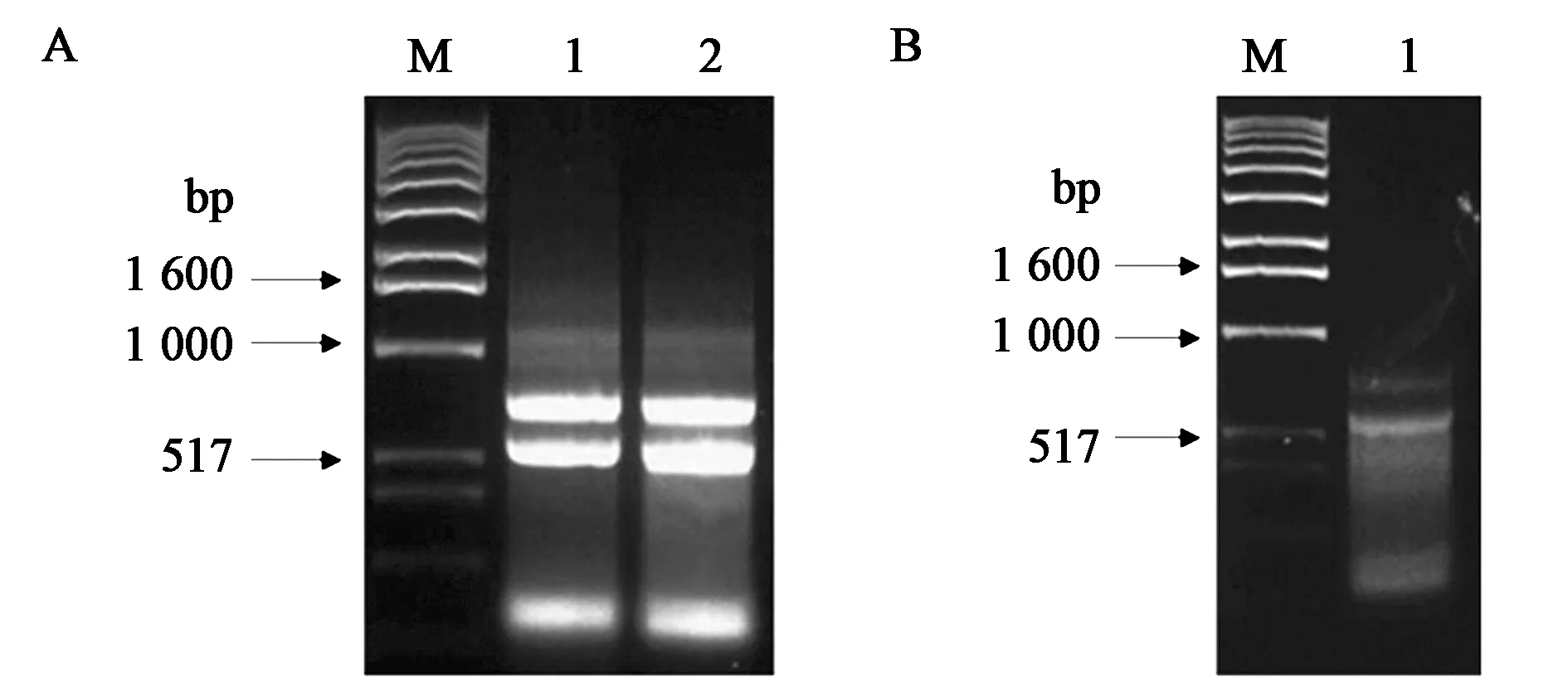
图5 RNA提取及去帽处理Fig.5 RNA extraction and decapped RNA.注:A:GS115 RNA;M:8 kb标准Marker;1~2:毕赤酵母GS115 RNA。B:GS115去帽RNA;M:8 kb标准Marker;1:毕赤酵母GS115去帽RNA。
为进一步提高PCR产物的特异性,以第一轮PCR产物为模板、引物为内部引物(ⅡF/ⅡR)进行第二轮PCR。PCR产物理论大小约为0.69 kb,这一结果经琼脂糖凝胶电泳进一步证实(图6C)。以上结果表明,利用嵌套PCR可获得目标片段。
2.6 转录起始位点确定
上述嵌套PCR产物连接至克隆载体pEASY-Blunt,通用引物M13F/M13R对转化子进行PCR验证(图7)。由于实验过程中RNA产生不同程度的降解,导致连接到测序载体的片段大小不一。
选取较大的PCR产物对应的转化子测序,测序结果使用Vector NIT软件进行序列比对(图8),其中LRA3为LRA3基因的启动子和编码区序列,其他序列均为测序所得。CRRT-PCR实验原理是将mRNA的首尾相连,mRNA的首个碱基即为转录起始位点而尾部则是polyA,即polyA后的第一碱基即为启动子上转录起始位点。在本研究中,polyA后面第一个碱基为G。但启动子上G的前一个碱基亦为A,故这个A是ployA还是转录起始位点无法确定。因此,转录起始位点可能为A或G。根据大量研究结果统计,转录起始位点通常为A[10],在本研究中认定A为PLRA3的转录起始位点。
2.7 PLRA3核心启动子及核心元件预测
转录起始位点(transcription start site,TSS),通常为转录的第一个位点,核心启动子在TSS指导下发挥正确转录起始的作用。核心启动子一般包括以TSS为中心的上、下游各40个核苷酸,还包含一些重要的核心启动子元件,典型例如TATA Box、起始子Initiator[11]。当然,不是所有的启动子都含有所有的核心启动元件,甚至一些启动子没有核心启动子元件[12]。通过Promoter Scan在线分析PLRA3的核心启动子及核心启动子元件(图9),预测PLRA3核心启动子区为-256 bp~-6 bp,得分为69.20,TATA Box位于-32 bp~-36 bp,未预测到其他元件,此结果与之前研究的酵母核心启动子中唯一的保守基序是TATA Box结论一致[13]。

图6 CRRT-PCR引物示意图及嵌套PCR琼脂糖电泳Fig.6 The schematic diagram of CRRT-PCR primers and agarose gel of nested PCR.注:A:嵌套引物位置示意图。B:第一轮嵌套PCR产物;M:8 kb标准Marker;1~5号:温度梯度PCR产物。C:第二轮嵌套PCR产物;M:8 kb标准Marker;1~2:PCR产物。
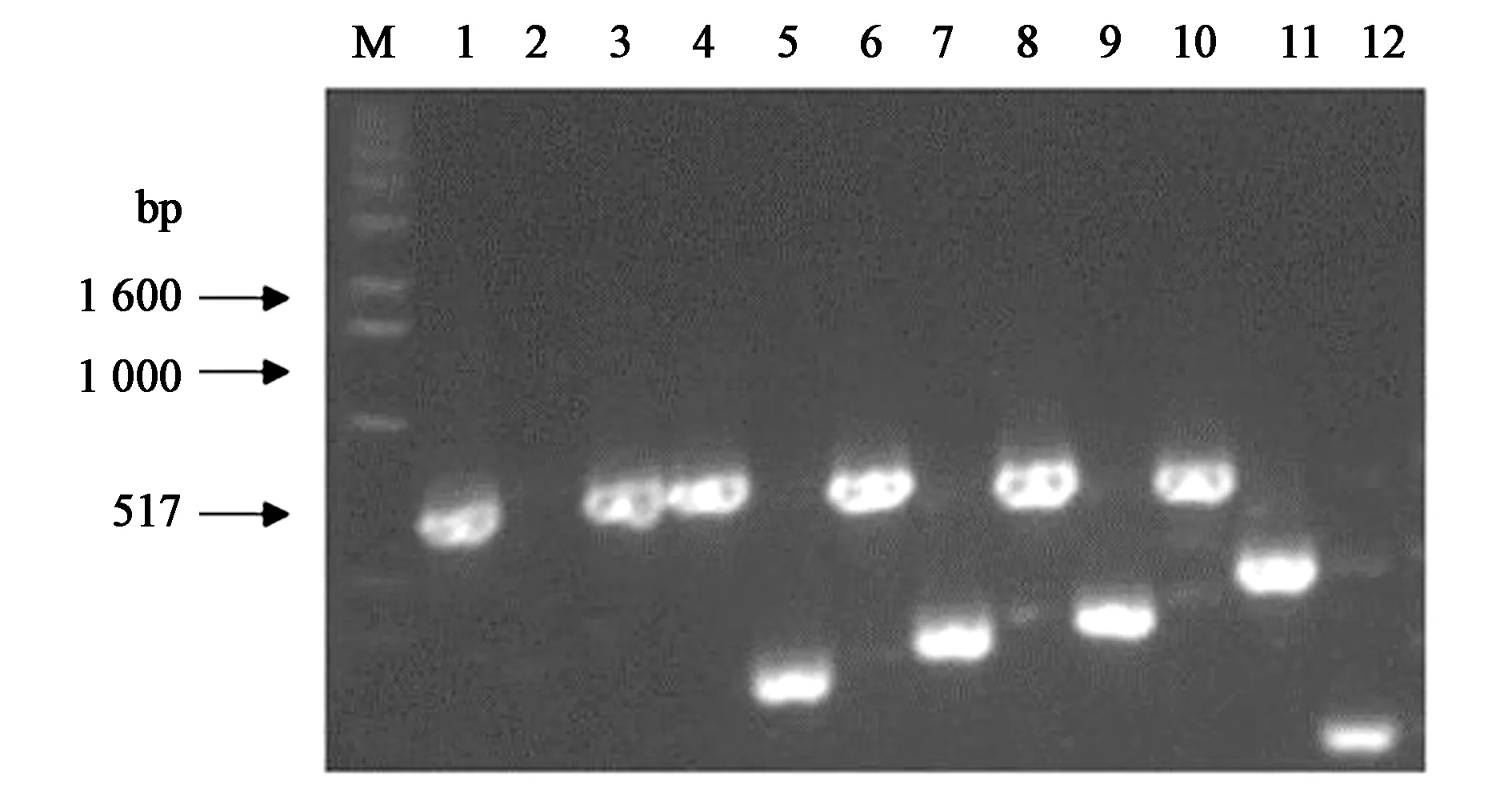
图7 PCR鉴定阳性转化子Fig.7 The PCR identification of transformants.注:M:8 kb标准Marker; 1~12:转化子。
2.8 PLRA3顺式作用元件预测
转录因子通过与启动子区域中的顺式元件相互作用,产生不同转录效应,基因启动子含有多种顺式作用元件,受到多个途径的调节[14]。CpG岛通常是一段大于200 bp、GC含量不小于50%、同时CpG含量与期望含量的比值不小于0.6的区域[15]。CpG岛主要存在于启动子区,通常情况其为非甲基化状态,一旦受到调控发生甲基化会抑制转录因子结合,最终造成基因沉默[16]。利用CpG Finder和EMBOSS Cpgplot在线分析PLRA3的CpG岛存在情况,预测表明PLRA3不存在CpG岛,即PLRA3不受甲基化调控。利用Nsite预测PLRA3的顺式作用元件,同时结合Promoter Scan结果,将二者重合结果部分列出,具体见表2。

图8 多序列比对结果Fig.8 The result of multiple sequences alignment.注:灰色阴影区为polyA;字体加粗的A为LRA3基因的转录起始位点;黑框为LRA3基因的起始密码子。

图9 LRA3基因图谱及PLRA3启动子序列Fig.9 The map of the LRA3 gene and the promoter PLRA3 sequence.注:下划线为核心启动子;黑框为TATA Box;箭头指示为转录起始位点。

表2 PLRA3顺式作用元件预测Table 2 The prediction of cis-acting elements of PLRA3.
3 讨论
启动子是表达系统的关键元件之一,在重组蛋白表达中扮演着重要角色。目前,应用于毕赤酵母表达系统的诱导型启动子可分为天然启动子和合成启动子。其中,天然启动子来源于一些在特定条件下表达谱或转录谱波动较大的基因,天然启动子往往存在一定缺陷,如转录强度不足和基础转录高等,需要进行理性改造。在对顺式作用元件研究较为清楚的基础上,可通过控制关键顺式元件位置、数量和间距构建合成启动子[19]。Vogl等[20]通过向启动子最小共有序列掺入常见的转录因子结合位点基序构建合成核心启动子,并在此基础上添加PAOX1上游序列得到合成启动子,其在甲醇诱导外源蛋白表达时达到野生型PAOX1核心启动子的10%。Portela等[21]从头合成核心启动子并与毕赤酵母基因AOX1上游调控区融合,其驱动外源蛋白表达水平最高可达野生型PAOX1的70.6%。显然,合成启动子是在揭示了天然启动子上关键调控元件的基础上对天然启动子进行改良和优化,从而避免天然启动子存在的一些弊端。
本实验室挖掘的鼠李糖强诱导型启动子PLRA3可以实现重组蛋白在毕赤酵母中的高效表达,但表达强度仍有提高的潜力以显著提高重组蛋白表达水平;该启动子在无鼠李糖诱导时还有较高的基础转录,重组蛋白存在渗漏表达,在表达对宿主有毒性作用的重组蛋白时效果不够理想[6~8]。因此,需要揭示该启动子上关键元件以进行理性改良。本研究通过敲除和回补实验证实了LRA3参与毕赤酵母鼠李糖代谢途径,并利用CRRT-PCR方法确定了转录起始位点A,同时预测了核心启动子、转录重要元件TATA Box及可能的顺式作用元件。在后期的研究中,将利用这些信息在启动子关键位点进行突变,从中筛选转录强度提升而基础转录强度降低的突变体。
