煤焦油加氢过程中NiWP/Al2O3催化剂失活研究*
2019-07-31吕木兰岑建孟夏芝香方梦祥
吕木兰 马 帅 岑建孟 夏芝香 方梦祥
0 引 言
中低温煤焦油催化加氢制取燃料油技术的应用,极大减轻了早期煤焦油直接燃烧带来的资源浪费和环境污染等问题。随着人们的环保意识增强,环保法规要求也逐渐提高,其中车用汽柴油国Ⅴ标准要求S含量不能高于10mg/kg,芳烃含量不能超过40%(质量分数,下同),多环芳烃不能超过11%;而国Ⅵ标准对芳烃的要求提高,芳烃含量不能超过35%,多环芳烃不能超过7%。可见,研制更高活性的加氢催化剂成为煤焦油催化加氢制取燃料油的一项重要内容[1-2]。研究表明,添加了P助剂的NiWP/Al2O3催化剂对煤焦油加氢脱S,N和O杂原子,芳烃饱和的综合效果较好[3-4]。唐巍[5]用固定床反应器评价了不同P含量的NiWP/Al2O3催化剂对煤焦油混合模化物的加氢性能,并与金属磷化物催化剂和商用催化剂(MoNiW/Al2O3)的加氢活性比较,结果发现,添加0.8%的NiWP/Al2O3催化剂性能最优。相比之下,Ni2P催化剂的加氢脱氧活性太低,商用催化剂的加氢脱硫和加氢脱氮活性均较低,NiWP-0.8催化剂具有优异的杂原子脱除活性,但芳烃加氢活性略低。张明伟等[6]采用P改性NiW/γ-Al2O3催化剂在固定床加氢反应装置进行低温煤焦油的催化评价,结果表明,助剂P的加入可以调变催化剂的孔径和酸性分布,活性组分在催化剂表面分布均匀;P的加入量为0.8%时,产品油中S和N的脱除率均达到97%以上。由此可见,NiWP/Al2O3催化剂的确是比较有前景的煤焦油加氢催化剂,但在反应过程中活性很快下降,加氢效果大打折扣,这限制了该催化剂的工业应用。对NiWP/Al2O3煤焦油加氢催化剂活性的研究比较多,而对其失活分析的研究较鲜见。催化剂失活研究较多的有柴油[7-8]、重油[9-11]和渣油[12]等加氢催化剂,而煤焦油与它们性质有差异。因此,本实验对NiWP/Al2O3催化剂的失活分析就很有必要。一般来说,催化剂失活的原因主要有积碳、烧结和金属中毒等[13-15],研究NiWP/Al2O3催化剂初期失活原因,对于延缓催化剂失活速率以及提高加氢活性具有现实应用意义。
本实验以活性氧化铝为载体,采用等体积浸渍法制备NiWP/Al2O3催化剂,并在实验室固定床加氢装置对催化剂进行不同时长的初期催化反应。通过采用N2吸附-脱附、XRD、元素分析、SEM、TPOMS、TG、FTIR、XPS、GC-MS等方法对催化剂反应前后的物化性质进行表征,考察催化剂初期快速失活的原因。
1 实验部分
1.1 催化剂的制备
采用等体积浸渍法制备NiWP/Al2O3催化剂。催化剂载体为商用活性氧化铝(国药集团化学试剂有限公司),浸渍液原料为硝酸镍(国药集团化学试剂有限公司,AR≥98%)、偏钨酸铵(麦克林生化科技有限公司,AR≥99.5%)、磷酸(国药集团化学试剂有限公司,AR≥85%)和去离子水。
操作流程为:将活性氧化铝压片、研磨、筛分,得到粒径为380μm~830μm的颗粒,110℃烘干2h;将一定量的硝酸镍、偏钨酸铵和磷酸配成溶液,室温下浸渍载体12h,接着120℃烘干6h,然后置于马弗炉中500℃焙烧4h,得到NiWP/Al2O3催化剂,其中 W,Ni,P的质量分数分别为30%,6%,0.8%。
1.2 催化剂的评价
催化剂性能的评价在固定床加氢微反装置上进行。等体积浸渍法得到的催化剂活性金属以氧化物状态存在,在加氢反应之前需进行预硫化活化处理,硫化液为5%CS2+95%环己烷的混合溶液,硫化条件为:硫化压力3MPa,硫化温度360℃,氢液体积比200∶1,体积空速10h-1。

表1 煤焦油馏分油模化物组成(%*)Table 1 Composition of coal tar distillate oil model compound(%* )
催化加氢反应条件为:反应压力6MPa,反应温度360℃,氢油体积比1 000∶1,体积空速1h-1。反应液是煤焦油馏分油模化物,由低温煤焦油中含量较高的8种代表性物质配制而成,其含量组成见表1。反应前三个小时的产物舍弃,之后每小时取一次样用于分析,反应原溶液及反应产物的S和N含量用江苏科苑有限公司KY3000SN型硫氮分析仪测定,反应物和产物的各组分含量用气相色谱(安捷伦科技有限公司)测定。硫化及反应后的催化剂从反应器中卸出后,用丙酮清洗表面残留的反应液,烘干,待分析表征用。氧化态催化剂、预硫化催化剂及反应了14h,20h,26h得到的催化剂分别命名为C0,C1,C14,C20,C26。
1.3 催化剂的表征
本实验采用美国麦克仪器公司生产的ASAP2460型物理化学吸附仪对催化剂的比表面积、孔体积和孔径分布进行分析。采用BET(Brunauer-Emmett-Teller)方法计算比表面积,采用BJH(Barrett-Joyner-Halenda)方法分析孔径分布。
采用荷兰 PANalytical B.V.公 司 的 X-pert Powder型X射线衍射仪对催化剂活性相结构进行XRD分析。仪器参数:Cu靶Kα射线,管电压为40 kV,管电流为15mA,扫描范围为5°~90°。
采用德国Vario MAX cube元素分析仪测定催化剂的C,H,S,N元素的质量分数。
采用日本日立公司的SU8010场发射扫描电镜分析加氢反应前后催化剂的表面形貌。
采用美国麦克仪器公司生产的AutoChem II2920型程序升温化学吸附分析仪与质谱仪联用进行TPO-MS表征,分析催化剂样品中积碳和硫等物质的氧化温度及其赋存状态。
采用德国耐驰仪器公司生产的TG209F3型热重分析仪分析反应后催化剂的失重过程。分析条件为:空气气氛,流速为100mL/min,温度范围为50℃~800℃,升温速率为5℃/min。
采用美国尼高力iS50傅氏转换红外线光谱仪测定催化剂样品中存在的有机官能团,为确定催化剂表面积碳的结构提供基础。测定条件:红外波数扫描范围为4 000cm-1~40cm-1,分辨率为0.2 cm-1,KBr压片,每分钟扫描16次。
采用日本岛津250XI PHI型X射线光电子能谱仪(XPS)测定催化剂表面含有的元素及同种元素的不同存在形式,用来测定催化剂表面C元素所处的形态及含量分布。
采用安捷伦7890B-5977A气相色谱质谱联用仪(GC-MS)分析反应后催化剂表面积碳的组成。用二氯甲烷提取催化剂表面的积碳,具体方法为:取2g反应了14h的催化剂于干净的烧杯中,加入20 mL二氯甲烷,静置0.5h,然后超声振荡1h,过滤,再将滤液加热浓缩至2mL~3mL。用气相色谱质谱联用仪分析提取液,以确定积碳组成。
2 结果与讨论
2.1 催化剂的活性评价
在固定床加氢装置上,进行NiWP/Al2O3催化剂催化煤焦油混合模化物不同时长的加氢反应。催化剂的反应活性以反应液的加氢脱硫、加氢脱氮、加氢脱氧和加氢脱芳烃活性作为指标进行评价。催化剂反应活性变化见图1。由图1可知,加氢反应活性最高时,S原子的脱除率在96%以上,N原子的脱除率达到94%以上。随着反应时间延长,二苯并噻吩的转化率变化不大,反应26h也能达到94%左右;而喹啉的转化率大概在反应进行11h时开始大幅下降,几个小时以后就下降到80%以下,甚至降到70%,反应产物溶液也由澄清透明状变得浑浊;邻甲酚的转化率稍有下降,但变化不大;芳烃中1-甲基萘和菲的转化率很高,且在反应时间内变化很小,联苯和邻二甲苯的转化率相对较小,随着反应进行,转化率明显下降。结果表明,NiWP/Al2O3催化剂对S,N,O杂原子的脱除率高,但反应活性快速下降,特别是加氢脱氮活性。
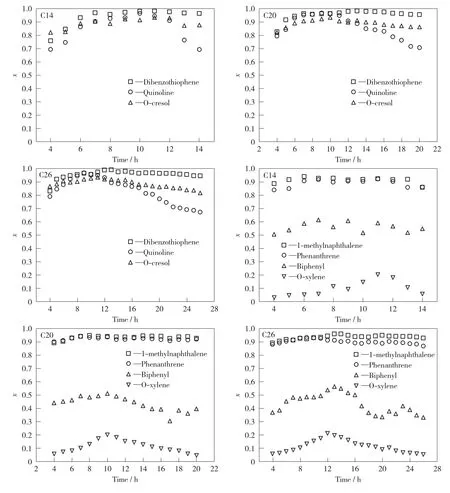
图1 模化物加氢反应的加氢脱硫、加氢脱氮、加氢脱氧和脱芳烃活性随时间的变化情况Fig.1 Hydrodesulfurization,hydrodenitrogenation,hydrodeoxygenation and hydrodearomatization activities of model compound hydrogenation reaction with time
2.2 催化剂表征
2.2.1 XRD表征结果
用Jade 6.5软件分析不同催化剂的XRD特征峰。图2所示为氧化态、硫化态及反应不同时间后催化剂的XRD谱。由图2可知,氧化态催化剂在2θ=67.2°处出现Al2O3晶体的特征衍射峰,在2θ=23.6°和33.5°处分别出现了 WO3的特征衍射峰,未见Ni和P氧化物的特征衍射峰,说明Ni和P均匀分布在金属表面,而活性金属W由于含量较高,在载体表面的分布没有那么均匀,出现少许金属团聚现象。催化剂硫化后,WO3反应生成 WS2,因此图2中出现了WS2的特征衍射峰。催化剂进行了不同时长的催化加氢反应后的XRD谱与反应前的谱基本一致,没有出现新的晶相。可见,反应前后催化剂的物相结构没有变化,该反应温度下没有出现催化剂烧结现象,说明催化剂初期失活不是高温烧结而导致的。

图2 不同催化剂的XRD谱Fig.2 XRD spectra of different catalysts
2.2.2 N2吸附-脱附表征结果
反应前后催化剂的比表面积及孔径分布情况分别如表2和图3所示。由表2可知,氧化态催化剂经预硫化后比表面积略有下降,总孔容减小,平均孔径有所增大,催化剂硫化处理会略微改变其孔结构。反应后催化剂的比表面积、总孔容和平均孔径与硫化后的相比都大大降低。结合图3可以看出,反应后催化剂的孔径分布曲线往孔径减小的方向移动,孔径为2nm~20nm范围的孔显著减少。ANCHEYTA et al[9]的研究发现,柴油催化加氢反应初期易发生结焦,沉积在催化剂表面,覆盖表面的活性中心且堵塞孔道,使催化剂活性下降。由此可推测本研究催化剂反应初期活性快速下降的原因是,催化剂表面生成积碳,覆盖了表面的部分反应活性位点,而且孔径的减小使反应液中的大分子无法进入孔道反应,从而催化剂的反应活性下降。
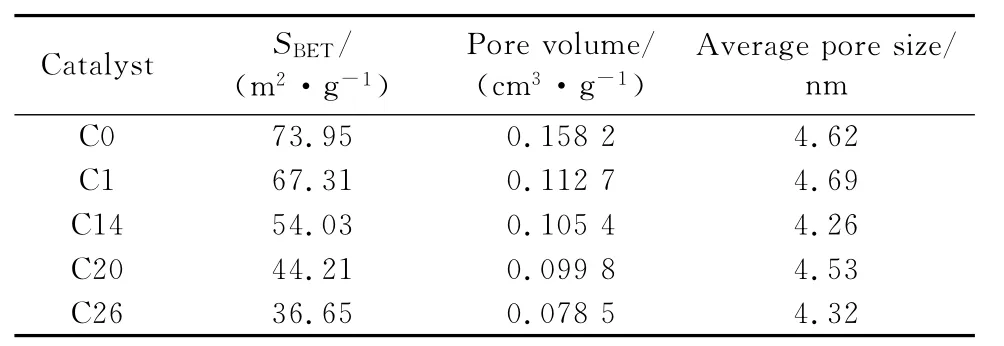
表2 不同催化剂的孔结构参数Table 2 Pore structure parameters of different catalysts

图3 不同催化剂的孔径分布Fig.3 Pore size distribution of different catalysts
2.2.3 SEM 表征结果
图4所示为不同催化剂的SEM照片。由图4可知,负载了活性金属后制得的催化剂表面仍有氧化铝载体的多孔结构且孔隙较大。硫化后催化剂表面颗粒相较有所变大,但是仍保持着原来的多孔结构。催化加氢反应后催化剂表面的孔道逐渐变小,孔口边缘有颗粒聚集,堵塞表面孔道,可能是积碳附着在催化剂表面,使反应活性位点变少,同时表面部分孔道的减小使反应物进入孔道进行反应变得困难,从而催化剂的活性下降。
2.2.4 元素分析
反应前后不同催化剂的元素分析见表3。由表3可知,反应了14h后的催化剂检测到3.99%的碳元素,而且随着反应时间增加,催化剂中碳元素含量明显增加,说明反应后催化剂出现结焦的情况。硫化处理后催化剂可以检测到较高的硫元素含量,此外,其数值随着反应进行也慢慢变大,可能催化剂沉积的部分积碳是由含硫化合物反应生成的。催化剂的氢元素和氮元素含量均较少,看不出明显变化规律。
2.2.5 TPO-MS表征结果
加氢反应后失活催化剂的TPO-MS谱如图5所示。由图5可知,水有两个信号峰,100℃的低温信号峰是催化剂失去表面水分导致的,而250℃~550℃的高温信号峰是催化剂上含氢物质发生氧化反应生成水的原因。在温度200℃~600℃范围内可检测到二氧化硫的存在,分别有350℃附近较强信号峰和500℃处较弱信号峰。文献[16]发现,在CoMo/Al2O3催化剂的空气氧化过程中,生成的二氧化硫峰值约在235℃和435℃处,第一个最大值主要来自于MoS2类硫化物的氧化,而第二个最大值是被氧化物/硫酸盐层覆盖的未反应的硫化物氧化产生。同时,文献[17]中NiMo/Al2O3催化剂的TPO-MS实验发现,二氧化硫在较高温度(478℃)附近产生,可能是吸附在样品或沉积在积碳中的含硫化合物的分解氧化产生的。依此类推,可以认为该催化剂的TPO-MS实验在低温区产生的二氧化硫,是由催化剂自身的硫化物氧化产生的,而高温区生成的二氧化硫可能是积碳中的硫产生的,峰的强弱与两类硫的实际含量接近。在400℃~600℃温度范围内可检测到碳的氧化物一氧化碳和二氧化碳的信号,其峰值均在500℃附近产生,可见催化剂的积碳在400℃才开始发生氧化反应,温度达到600℃时基本反应完全,这也为催化剂的氧化再生提供了温度参考。

图4 不同催化剂的SEM照片Fig.4 SEM photos of different catalysts
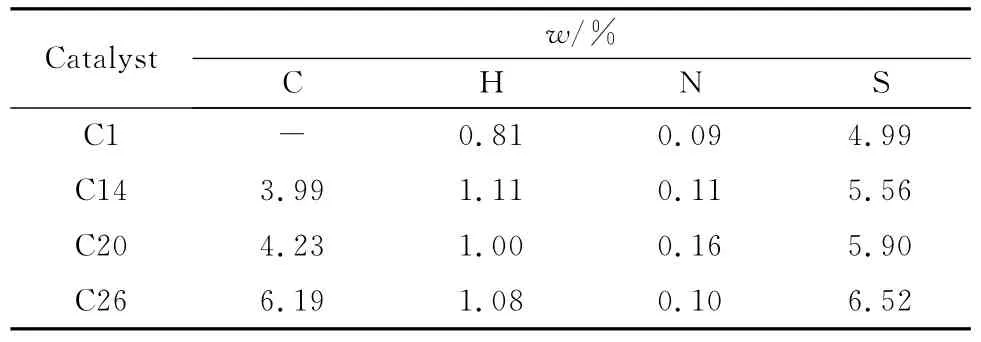
表3 不同催化剂的元素分析(ad)Table 3 Ultimate analysis of different catalysts(ad)
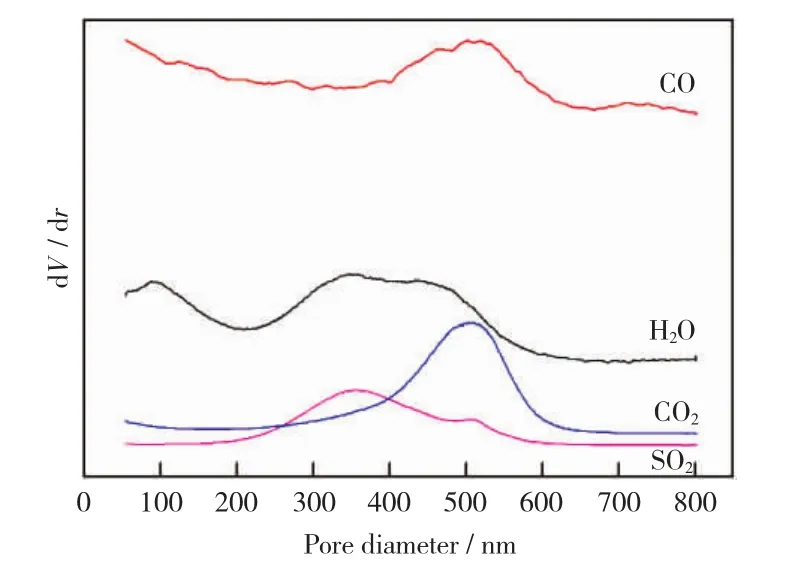
图5 失活催化剂的TPO-MS谱Fig.5 TPO-MS spectra of spent catalyst
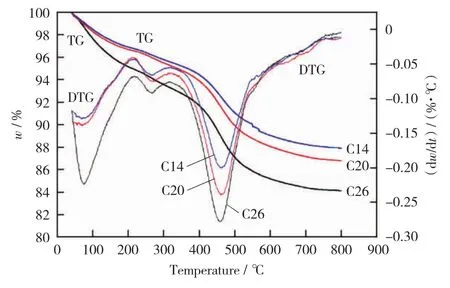
图6 反应后催化剂的TG-DTG曲线Fig.6 TG-DTG curves of the catalysts after the reaction
2.2.6 TG表征结果
图6所示为三种不同反应时长的催化剂失重曲线。由图6可知,三种催化剂的失重曲线的变化规律相同,均有三个失重峰,分别在温度为80℃,250℃,470℃左右处。结合反应后催化剂的TPO-MS分析结果可知,低温失重峰对应的是催化剂表面的物理吸附水分;200℃~350℃温度范围的失重对应着催化剂中少量含硫和氢物质发生氧化反应失去的重量;350℃~600℃温度范围的失重主要是因为高温氧化除去催化剂表面的积碳和部分含硫物质失去硫。三条曲线的不同在于随着反应时间增加,催化剂总失重量变大,可以看出中温段和高温段的失重增加,该范围失去的主要是硫元素和碳元素,说明催化剂含有的硫和碳元素含量增多,这与2.2.4节元素分析的结果一致。

图7 反应后不同催化剂的红外光谱Fig.7 Infrared spectra of different catalysts after the reaction
2.2.7 FTIR表征结果
前人已经在红外光谱测定积碳结构领域有了一定的研究,并总结归纳了红外光谱各吸收峰及其对应的基团等情况[18-19]。反应后催化剂的FTIR谱见图7。由图7可知,三种催化剂的红外光谱谱相差不大,出峰位置和峰的大小差不多。在吸收带3 500 cm-1~3 300cm-1区域内有一个峰形宽且钝的强吸收峰,主要为羟基(—OH)的伸缩振动;在3 000cm-1~2 800cm-1区域内有小的吸收峰,为烷烃C—H的伸缩振动,强度弱;在1 782cm-1处对应芳香烃的振动;在1 617cm-1处有一个较强的吸收峰,为具有—O—取代的芳烃C C的伸缩振动;在1 457cm-1处对应着烷烃C—H键的弯曲振动,强度弱;在1 100cm-1和560cm-1附近分别有一个较强的吸收峰,主要为杂原子取代,分别是C—N键和C—S键的伸缩振动;在872cm-1处对应着取代芳烃C—H的弯曲振动,强度很弱。说明在实验时间内,随着反应时间增加,催化剂表面生成积碳的基团种类没有改变。同时,从吸收峰的强度可以推测,积碳的成分主要为芳香烃,还含有少量脂肪烃。
2.2.8 XPS表征结果
2.2.7小节中对反应后催化剂的红外光谱分析初步确定了积碳结构含有的基本官能团,然后用X光电子射线能谱(XPS)分析方法进一步分析样品中存在的元素及化合物中同种元素的不同存在形态,反应后催化剂的XPS全谱如图8所示。由图8可以看出,三种样品的XPS全谱基本一致,说明在实验时间内,随着反应时间延长,催化剂所含元素一样,而且各元素谱峰的相对强度也没什么变化。组成催化剂的O,Al和W等元素的谱峰很明显。Ni元素含量较少,能看到较弱的峰,而含量更少的P元素基本检测不到。此外,样品中还含有较多的非金属元素C和少量的S元素,N元素因为量少基本检测不到,这与元素分析的结果相符。

图8 反应后不同催化剂的XPS谱Fig.8 XPS spectra of different catalysts after reaction
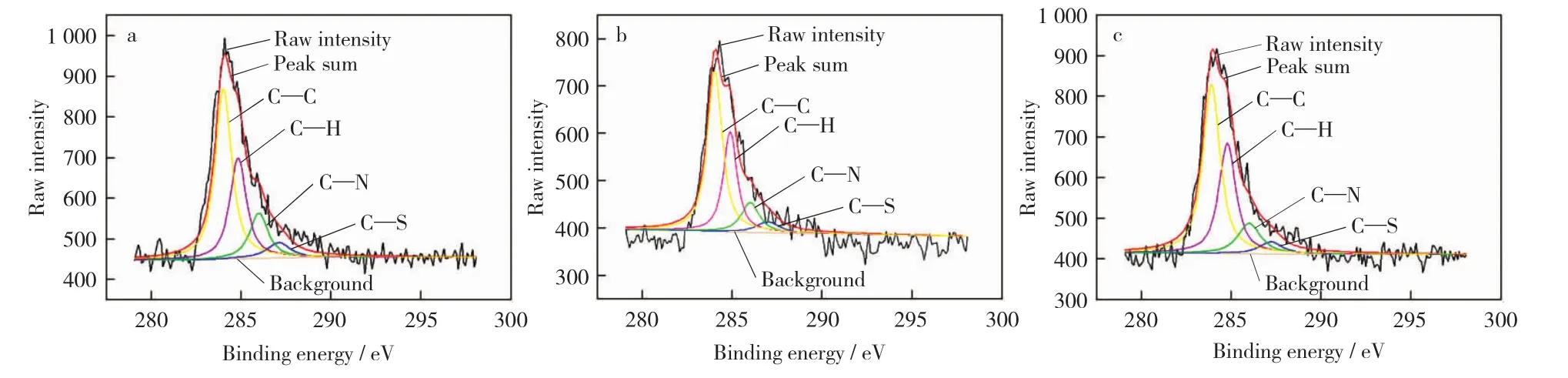
图9 不同催化剂的C1s分谱Fig.9 Different catalysts samples’C1s peak processing
为分析催化剂生成积碳的结构,用XPSPEAK41软件对C1s谱进行分峰拟合,确定积碳中C的存在形态,结果如图9所示。三种催化剂样品C1s分峰拟合结果接近,可得到4个不同能量的峰[19-20],分别是(287.00±0.2)eV 归属于C—S键的特征峰,(286.00±0.1)eV归属于C—N键的特征峰,(284.80±0.1)eV归属于C—H 键的特征峰,(284.00±0.1)eV归属于芳香化C—C键的特征峰,其中芳香化C—C键的峰面积明显比其他的大。红外光谱中强度较大的芳香化 C C键受到测试方法的限制,所以在XPS中未分辨出[20]。
由催化剂的XPS分析结果可知,催化剂表面积碳主要是含有杂原子S元素和N元素的芳香型烃类。
2.2.9 GC-MS分析结果
气相色谱质谱联用(GC-MS)分析结果如图10所示。表4所示为图10中几个主要的峰代表的物质。由表4可以看出,这几种物质包含在反应物或者反应物加氢过程的中间产物之中,含硫的烷烃可能是正十六烷与二苯并噻吩脱除的硫原子或者自由基结合而成。
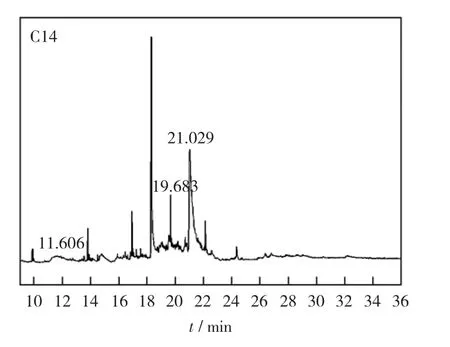
图10 催化剂的GC-MS分析色谱Fig.10 Chromatogram of GC-MS analysis of catalyst

表4 色谱峰的归属Table 4 Assignment of chromatography peaks
3 结 论
1)NiWP/Al2O3催化剂的加氢脱硫、加氢脱氮和加氢脱氧活性均较高,但是加氢脱氮活性在反应进行十个多小时就开始显著下降。相比之下,催化剂的加氢脱硫活性更好、更稳定,加氢脱氧活性也比较稳定。
2)加氢反应前后催化剂的晶相结构基本相同,没有高温导致的活性相烧结现象。但是反应后催化剂的比表面积、总孔容和平均孔径下降,催化剂上出现C元素,随着反应时间增长,C元素含量增加。由此可以认为,催化剂在反应初期快速失活是其表面生成积碳所致。
3)反应后催化剂除了其本身含有的物质,还有较多的C元素和少量的S和N元素,催化剂表面生成的积碳主要含有芳香烃,还含有少量脂肪烃,其中部分烃类有S和N杂原子。积碳组成有反应物,也有反应物加氢的中间产物。
