正交试验优选藤龙补中方水提工艺*
2019-07-05许聪聪王晓晗梁惠芬安红梅浦益琼
许聪聪 ,王晓晗 ,梁惠芬 ,林 鹭 ,安红梅 ,胡 兵 ,浦益琼 ,张 彤
(1.上海中医药大学教学实验中心,上海 201203; 2.上海中医药大学中药学院,上海 201203;3.上海中医药大学附属龙华医院,上海 201203)
藤龙补中方系上海中医药大学附属龙华医院的临床验方,主要用于治疗大肠癌、胃癌、胰腺癌等恶性肿瘤,由藤梨根、龙葵、蛇莓、白术、茯苓、薏苡仁、槲寄生、半枝莲等中药组方[1]。目前,藤龙补中汤配合手术、化学治疗(简称化疗)等方式联合治疗大肠癌,疗效明显[2-4]。藤龙补中汤可促进结肠癌细胞的衰老,抑制大肠癌细胞的转移,降低化疗不良反应,促进大肠癌患者胃肠道功能的恢复[5-7]。在确保原方疗效的同时,通过优化提取工艺可制备成固体制剂。本研究中以中医药理论为指导原则,在不改变提取溶剂的基础上,以浸膏得率、野黄芩苷、对羟基桂皮酸和多糖含量为考察指标,对藤龙补中方的水提工艺进行优选,为复方颗粒剂的研究提供样品和技术积累。现报道如下。
1 仪器与试药
仪器:1200型高效液相色谱仪(美国安捷伦公司);BS-124S型电子天平(德国赛多利斯公司);101-2-5型电热恒温鼓风干燥箱(上海跃进医疗器械厂);XS105DU型电子天平(瑞士梅特勒-托利多公司);RE-52AA型旋转蒸发仪(上海亚荣生化仪器厂);HHS型电热恒温水浴锅(上海博讯实业有限公司医疗设备厂)。
试药:藤梨根、龙葵、蛇莓、白术、茯苓、薏苡仁、槲寄生、半枝莲(上海康桥中药饮片有限公司,批号分别为161011,161223,160111,160818,160823,170117,160624,161223),经上海中医药大学中药学院张红梅博士鉴定为正品,均符合2015年版《中国药典(一部)》相关要求;D-无水葡萄糖对照品(批号为110833-201506),野黄芩苷对照品(批号为110842-201709),均购自中国食品药品检定研究院;对羟基桂皮酸对照品(上海誉恩生物科技有限公司,批号为7400-08-0);甲醇(批号为 R6AG1H)、乙腈(批号为 S8HA1H)均为色谱纯(Honeywell公司),其他试剂均为分析纯(国药集团化学试剂有限公司)。

图1 高效液相色谱图
2 方法与结果
2.1 野黄芩苷含量测定[8]
2.1.1 色谱条件及系统适用性试验
色谱柱:DiamonsilTMPlus C18柱(250 mm×4.6 mm,5 μm);流动相:甲醇 -0.1% 磷酸溶液(39 ∶61);流速:1.0 mL/min;检测波长:335 nm;柱温:25 ℃;进样量:10 μL。在此色谱条件下的色谱图见图1。
2.1.2 溶液制备
取野黄芩苷对照品适量,精密称定,加甲醇制成质量浓度为1.028 g/L的对照品贮备液。取浸膏粉约0.25 g,精密称定,置具塞锥形瓶中,加入50%甲醇溶液100 mL,超声10 min,摇匀,过滤,即得供试品溶液。
2.1.3 方法学考察
线性关系考察:取对照品贮备液适量,加甲醇制得质量浓度分别为 8.224,20.56,51.4,128.5,257,514,1 028 μg/mL的系列对照品溶液,按拟订色谱条件进样测定,以质量浓度为横坐标(X)、峰面积为纵坐标(Y)进行线性回归,得回归方程Y=30.919X-0.613 36,r=0.9995(n=7)。结果表明,野黄芩苷质量浓度在8.224~1028g/L范围内与峰面积线性关系良好。
精密度试验:分别精密吸取质量浓度为20.56,128.5,514 μg/mL 对照品溶液,各 10 μL,注入液相色谱仪,连续测定6次。结果的RSD分别为1.31%,0.69%,0.23%(n=6),表明仪器精密度良好。
稳定性试验:取浸膏,平行测定6份,依法制备供试品溶液,按拟订色谱条件进样测定。结果的RSD为1.44%(n=6),表明供试品溶液在24 h内基本稳定。
重复性试验:取浸膏,依法制备供试品溶液,按拟订色谱条件进样测定。结果平均含量为1.42%,RSD为1.53%(n=6),表明方法重复性良好。
加样回收试验:取已知含量的浸膏9份,分别加入相当于样品含量50%,100%,150%的对照品,依法制备供试品溶液,按拟订色谱条件测定。结果见表1。
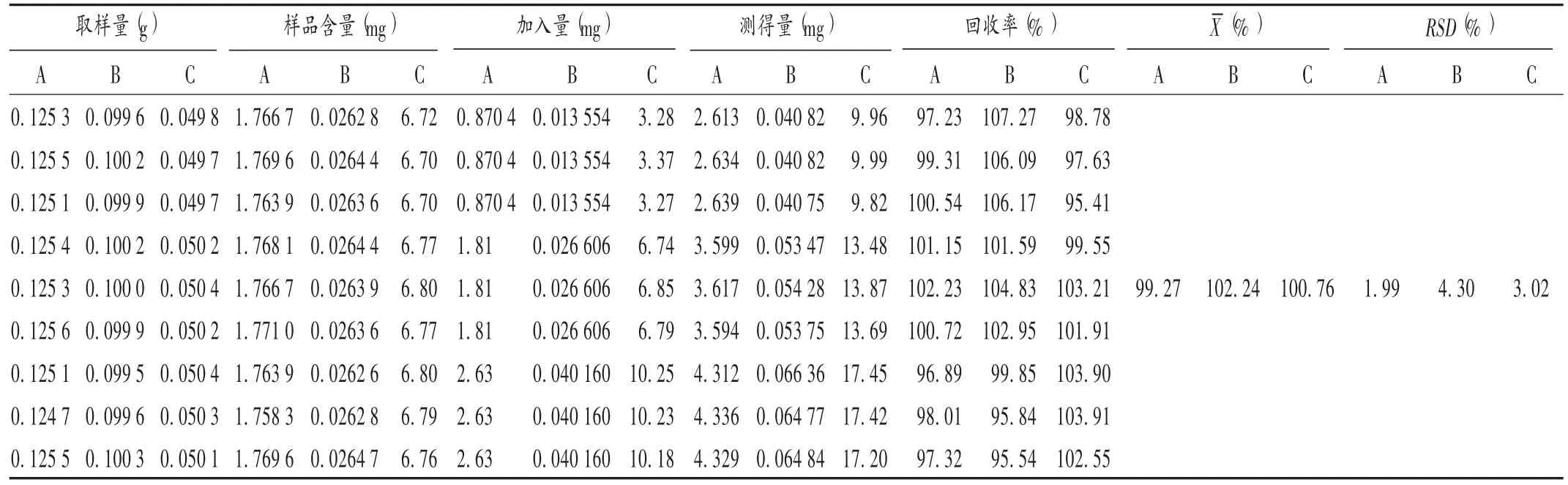
表1 加样回收试验结果(n=3)
2.2 对羟基桂皮酸含量测定[9-10]
2.2.1 色谱条件及系统适用性试验
色谱柱:UltimateXB-C18柱(250mm×4.6mm,5μm);流动相:乙腈 -0.2%醋酸溶液(14∶86);流速:1.0mL/min;检测波长:300 nm;柱温:25 ℃ ;进样量:10 μL。在此色谱条件下的色谱图见图1。
2.2.2 溶液制备
取对羟基桂皮酸对照品适量,精密称定,加甲醇制成质量浓度为1.04 g/L的对照品贮备液。取浸膏粉约0.2 g,精密称定,置具塞锥形瓶中,加入50%甲醇溶液25 mL,超声20 min,摇匀,过滤,即得供试品溶液。
2.2.3 方法学考察
线性关系考察:取对羟基桂皮酸对照品贮备液适量,加甲醇制成质量浓度分别为0.784 375,1.568 75,3.137 5,6.275,12.55,25.1,50.2 μg/mL 的系列对照品溶液,按拟订色谱条件进样测定,以质量浓度为横坐标(X)、峰面积为纵坐标(Y)进行线性回归,得回归方程Y=70.579X-17.853,r=0.999 9(n=7)。结果表明,对羟基桂皮酸质量浓度在0.784 375~50.2 μg/mL范围内与峰面积线性关系良好。
精密度试验:分别精密吸取质量浓度为1.568 75,6.275,25.1 μg/mL 的对照品溶液,各 10 μL,注入液相色谱仪,连续测定6次。结果的RSD分别为1.18%,1.17%,0.42%(n=6),表明仪器精密度良好。
稳定性试验:取同一供试品溶液,按拟订色谱条件于24 h内间隔一定时间进样6次。结果的RSD为2.00%(n=6),表明供试品溶液在24 h内基本稳定。
重复性试验:取浸膏,平行6份,依法制备供试品溶液,按拟订色谱条件进样测定。结果平均含量为0.026 4%,RSD为1.52%(n=6),表明方法重复性良好。
加样回收试验:取已知含量的浸膏9份,分别加入相当于样品含量50%,100%,150%的对照品,依法制备供试品溶液,按拟订色谱条件进样测定。结果见表1。
2.3 多糖含量测定[6,11-12]
2.3.1 溶液制备
取干燥至恒定质量的D-无水葡萄糖对照品8.77mg,置10 mL容量瓶中,以蒸馏水定容至刻度,配制成质量浓度为877 μg/mL的对照品贮备液。取浸膏粉约0.1 g,精密称定,加5 mL水使其完全溶解,加入95%乙醇,搅拌均匀,4℃冰箱静置过夜,过滤,沉淀用95%乙醇洗涤3次,干燥。将干燥后的沉淀全部溶解于100 mL容量瓶中,加水至刻度,摇匀,过滤,即得供试品溶液。
2.3.2 测定方法
取供试品溶液1.0 mL,置试管中,加入5%苯酚溶液1.0 mL,摇匀,迅速加入浓硫酸5.0 mL,混匀,沸水浴中加热15 min后,迅速冷却至室温,于487 nm波长处测定吸光度,计算多糖含量。
2.3.3 方法学考察
线性关系考察:取葡萄糖对照品贮备液适量,加水制备成质量浓度 分别 为 19.294,29.818,38.588,49.989,70.16,79.83 μg/mL 的系列对照品溶液,精密吸取1.0 mL,加入5%苯酚溶液1.0 mL,摇匀,迅速加入浓硫酸5.0 mL,混匀,沸水浴中加热15 min后,迅速冷却至室温,以相应试剂为空白,于487 nm波长处测定吸光度(A)值,以 A(Y)为纵坐标、质量浓度(X)为横坐标进行线性回归,得回归方程Y=9.9X+12.9,r=0.999 7(n=6)。结果表明,D-无水葡萄糖质量浓度在19.294~79.83 μg/mL范围内与峰面积线性关系良好。
精密度试验:分别称取质量浓度为29.818,49.989,79.83 μg/mL的对照品溶液适量,按拟订色谱条件测定吸光度,连续测定6次。结果的RSD分别为0.11%,0.15%,0.03%(n=6),表明仪器精密度良好。
稳定性试验:取同一供试品溶液,按拟订色谱条件于24 h内间隔一定时间测定6次。结果的RSD为2.70%(n=6),表明供试品溶液在24 h内稳定性良好。
重复性试验:取浸膏,依法制备供试品溶液,按拟订色谱条件测定吸光度。结果平均含量为13.49%,RSD为2.47%(n=6),表明方法重复性良好。
加样回收试验:取已知含量的浸膏9份,分别加入相当于样品含量50%,100%,150%的对照品,依法制备供试品溶液,按拟订色谱条件测定。结果见表1。
2.4 水提工艺优选
选择浸泡时间(因素A)、加水量(因素B)和提取时间(因素C)为考察因素[13],以浸膏得率(指标Ⅰ)、野黄芩苷(指标Ⅱ)、对羟基桂皮酸(指标Ⅲ)及多糖(指标Ⅳ)含量的综合评分为考察指标,权重系数设为0.25。称取处方量药材,按L9(34)正交表设计试验,试验安排及结果见表2,方差分析见表3。

表2 藤龙补中方水提工艺正交试验分析
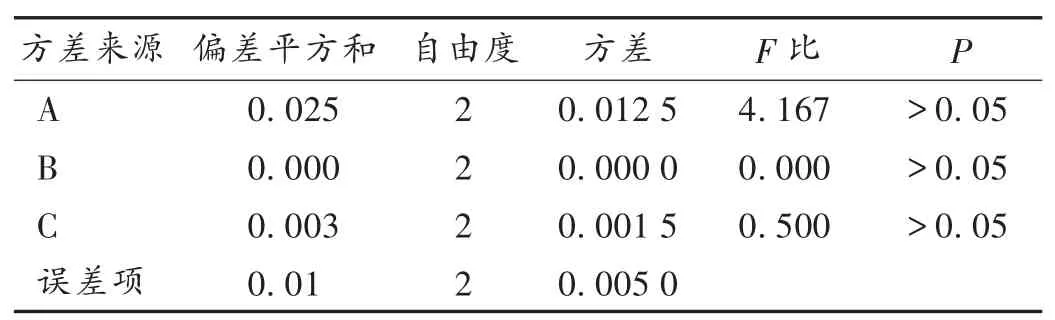
表3 水提工艺方差分析
由方差分析可知,因素A(温度)、因素B(溶剂用量)和因素C(提取时间)均对结果的影响无显著性,因此采用直观分析结果。各因素的最佳搭配为A3B2C1,即采用浸泡0 h,溶剂用量(10+8)倍,提取时间2 h;为节约能源,溶剂用量不变,提取时间调整为1 h。
按优选的水提工艺提取,经3批验证试验,所得浸膏中野黄芩苷、对羟基桂皮酸和多糖含量分别为1.39%,0.027 9%和15.25%,RSD分别为 2.79%,0.67%和2.82%,浸膏得率为15.89%,RSD为1.78%(n=3),表明该水提工艺稳定、可行。
3 讨论
测定野黄芩苷含量时,曾分别考察了提取溶剂(50% ,70% ,100%甲醇)、溶剂用量(10,25,50,100 mL)及超声时间(10,20,30 min)对野黄芩苷提取率的影响。结果表明,当取样量为0.25 g,提取溶剂为50%甲醇100 mL,超声提取10 min,野黄芩苷提取率较高。
测定对羟基桂皮酸含量时,也曾分别考察了提取溶剂(50%,70% ,100% 甲醇)、溶剂用量(10,25,50 mL)及超声时间(10,20,30 min)对对羟基桂皮酸提取率的影响。当取样量为0.2 g,提取溶剂为50%甲醇50 mL,超声提取20 min,对羟基桂皮酸的提取率较高。
野黄芩苷为半枝莲的主要成分,在该成分的测定过程中,先采用与2015年版《中国药典(一部)》半枝莲药材项下含量测定方法一致的甲醇-水-醋酸(35∶61∶4,V∶V∶V)流动相进行测定,由于野黄芩苷色谱峰与相邻峰未分开,未达到要求。初步考虑该复方成分较复杂,可能是某类黄酮类化合物对其造成了影响,需重新调整流动相。现行药典中灯盏细辛含有野黄芩苷,以甲醇-0.1%磷酸溶液(40∶60,V∶V)为流动相,并根据具体情况将流动相调整为甲醇-0.1%磷酸溶液(39∶61,V∶V),可获得较好的色谱峰分离效果。
在建立了3种活性成分含量测定方法的基础上,以综合评分法优选该方的水提工艺。选定的3个指标成分中,野黄芩苷和多糖成分的抗肿瘤作用显著。半枝莲中的黄酮类化合物在体内外都有显著的消除自由基的作用[14],对人结肠癌细胞株SM480有抑制作用[15];此外,该复方水提物中含有大量多糖成分,也具有良好的抗肿瘤作用。其中,半枝莲多糖能抑制HepA小鼠肿瘤生长,降低H22肝癌小鼠瘤重[16],提高C26结肠癌小鼠的肿瘤组织凋亡蛋白Caspase-3和Caspase-9的活性发挥抗肿瘤作用[17];茯苓多糖对胃癌、乳腺癌、肝癌均能起到一定的抑制作用[18];藤梨根中的有效成分猕猴桃根多糖可通过抑制PCNA蛋白的过速增殖而抑制结肠癌和肝癌的增殖[19];龙葵多糖能显著抑制小鼠U14细胞的增殖,延长荷瘤小鼠的生存时间,是治疗宫颈癌的有效物质,龙葵中分离得到的糖蛋白类成分能抑制结肠癌细胞HT-29和HCT-116细胞株的增殖,提示该药中糖蛋白类成分可能是抗结肠癌的药效物质[20]。
