德尔卑沙门氏菌血清型分子检测靶点筛选及多重PCR检测方法的建立
2019-05-07翟立公杨剑婷李永泉王水平王俊颖
翟立公,杨剑婷,李永泉,王水平,王俊颖
(安徽科技学院 食品工程学院,安徽 滁州,233100)
沙门氏菌是一种重要的食源性致病菌,能引起肠胃炎、伤寒和败血症等。据统计,在美国被沙门氏菌感染人群达到120万人次,造成300多人的死亡[1]。有文献报道,沙门氏菌有2 600多个血清型,其中具有致病性的血清型主要集中在肠炎沙门氏菌种的1 400多个血清型内[2]。根据调查发现,沙门氏菌污染具有地域性和血清型的特点,而且由沙门氏菌引起的污染事件大多是由一种或几种血清型引起的[3-4]。陈玲等[5]对南方的七大类400份食品中沙门氏菌的污染情况进行调查,结果表明,检出率最高的血清型分别是德尔卑沙门氏菌(SalmonellaDerby, SD)和鼠伤寒沙门氏菌。在对合肥地区的沙门氏菌污染调查过程中共检测出沙门氏菌22株,其中SD占到45.6%[6]。在对意大利撒丁区的猪肉产品及屠宰环境的检测过程中发现,沙门氏菌的检出率为12.9%,其中主要的优势血清型包括S. Anatum(40%)、S. Derby(18.5%) 和S. Rissen(20%)[7-8]。近年来调查发现,在欧洲地区SD血清型是生猪和猪肉制品污染的主要血清型[8-9]。经研究表明,主要是由于该血清型对猪的空肠源IPEC-J2细胞侵染较强所致[10]。虽然SD没有沙门氏菌其他血清型的感染性强,但对于免疫力低下的人群包括婴幼儿或老人等SD感染的感染率较高。
SD血清型的检测主要利用国标GBT 4789.4—2010的传统培养的方法,该方法结果较为准确,但检测时间较长(5 d以上)、操作复杂(预增菌、选择性培养、显示培养、生化鉴定和血清型鉴定等步骤)、成本高,而且如果凝集反应迟缓或特异性差,将造成判断错误[2,11]。随着现代食品工业的发展,为了适应样品多、检验时间短和检验结果准确等要求,有研究者提出针对沙门氏菌血清型开发分子检测技术。因此,解决沙门氏菌血清型分子检测技术的核心问题是能否筛选到血清型特异性较好的检测靶点。比较基因组学(comparative genomics)是在基因水平上寻找生物体之间的差异。随着不同血清型沙门氏菌全基因组测序的完成,可以利用比较基因组学方法筛选到目的血清型的特异性基因片段,并以此为检测靶点建立相应的分子检测方法[3]。DAVID等[12]通过比较基因组学的方法成功筛选到了猪霍乱沙门氏菌和丙型副伤寒沙门氏菌的血清型特异性基因片段,并以此为模板设计引物,建立了能同时检测以上2个血清型的多重PCR检测体系。
本研究利用生物信息学和比较基因组学方法筛选SD血清型特异性基因,并以此为检测靶点设计引物,与沙门氏菌属特异性引物(139-141)建立SD检测的多重PCR检测体系,并对其特异性、灵敏度、抗干扰能力和重现性等方面进行评估,为食品样品中沙门氏菌血清型的快速检测提供技术支持。
1 材料与方法
1.1 材料与试剂
标准菌株:39株沙门氏菌(沙门氏菌31血清型)和18株非沙门氏菌;分离株:11株(沙门氏菌5血清型),菌株具体信息见表2。细菌基因组DNA提取试剂盒、 TaqDNA聚合酶和Marker DS2000,广州东盛科技有限公司;食品样品(猪肉、鸡肉和牛肉),市售。
1.2 仪器和设备
PCR仪,杭州博日科技有限公司;电泳仪,北京六一仪器有限公司;凝胶成像分析仪,上海培清科技有限公司;二级生物安全柜,济南鑫贝西生物技术有限公司;
1.3 方法
1.3.1 基因组DNA提取及浓度测定
将过夜培养后的菌液,根据EasyPure Genomic DNA Kit说明书操作方法和热裂解法[13]提取各菌液的基因组DNA,利用核酸浓度测定仪分析提取的基因组DNA的纯度和浓度,将质量较好的基因组DNA置于-20 ℃冰箱保存备用。
1.3.2 SD血清型特异性基因的筛选
在NCBI(national center for biotechnology information)的GenBank数据库中获取SD(CVM N42507)的基因组序列信息,用于后续血清型特异性靶点的发掘。利用BLASTn(https://blast.ncbi.nlm.nih.gov/Blast.cgi)系统对该血清型的每个编码基因进行比对,按照SD血清型内同源性较高,但与其他血清型不具同源性的原则筛选SD血清型分子检测的准特异性靶点。
根据已获得的准特异性基因为模板,按照引物设计原则利用Primer 6.0软件设计引物,并通过BLASTn分析引物的特异性,本研究所涉及的引物见表1。以实验室保存的微生物DNA为模板,进行PCR扩增(反应体系和扩增程序略),验证SD血清型检测引物的特异性,确定SD血清型分子检测靶点。

表1 候选引物及序列
1.3.3 SD血清型多重PCR检测体系的建立
将SD血清型特异性引物D1、D3与沙门氏菌属特异性引物139-141结合,优化反应体系。最终确定SD血清型检测多重PCR的反应体系为:10×Taq buffer 2.5 μL、Taq聚合酶0.2 U/μL、MgCl24 mmol/L、dNTP 0.45 mmol/L、引物D1 0.8 mol/L、引物D3 0.6 mol/L、引物139-141 0.4 mol/L、DNA模板1 μL和ddH2O 5.5 μL。PCR反应条件:94 ℃预变性8 min;30 个循环,每个循环94 ℃变性30 s,60 ℃退火40 s,72 ℃延伸60 s;最后72 ℃延伸5 min。PCR产物通过2%的琼脂糖凝胶进行电泳,观察结果。
1.3.4 SD检测体系的特异性和灵敏度评估
提取实验室保存的微生物基因组DNA为检测模板,其中SD菌株的基因组为阳性模板,其他微生物基因组DNA为阴性模板,利用SD血清型多重PCR反应体系进行扩增,验证该检测体系的特异性。
测定SD血清型基因组DNA浓度,用无菌水10倍梯度稀释,并将各稀释度取1 μL作为模板,利用SD血清型多重PCR检测体系进行扩增,验证DNA模板灵敏度。
利用平板菌落计数法获得过夜培养的SD菌液的初始浓度,再通过无菌生理盐水进行10倍梯度稀释,各稀释度分别取1 mL,提取基因组DNA进行PCR检测体系扩增,验证该体系检测的菌落灵敏度。
1.3.5 SD检测体系的抗干扰能力评估
超市购买的猪肉、鸡肉和牛肉,经过GB 4789.4—2010方法验证无沙门氏菌污染后,冰箱冻存待用。3种样品各取25 g在缓冲蛋白胨液体培养基中进行过夜培养,获得猪肉背景菌群增菌液、鸡肉背景菌群增菌液和牛肉背景菌群增菌液,并通过平板计数法得到以上3种增菌液的初始浓度,用无菌生理盐水进行梯度稀释。同时,将SD(CMCC50719)进行过夜培养,并测定菌液的初始浓度,用无菌生理盐水进行梯度稀释。将SD菌液和以上3种样品增菌液分别按照1∶1、1∶102、1∶104、和1∶106菌落浓度比例进行混合,提取混合液基因组DNA,PCR扩增,以无菌水为空白对照。
1.3.6 SD检测体系的人工污染试验
将超市购买的猪肉、鸡肉和牛肉样品,进行巴氏杀菌,每类样品各取6份(每份25 g),分别用1 mL不同浓度的SD菌液进行污染,置于LB液体培养基中,37 ℃增菌培养,增菌6 h后每隔2 h取1 mL增菌液,热裂解法提取基因组DNA作为模板,进行PCR检测。
将超市购买的猪肉、鸡肉和牛肉样品,进行巴氏杀菌,每份样品25 g,分别用1 mL不同浓度的SD菌液进行污染,每份样品分别利用沙门氏菌传统检测方法(GB 4789.4—2010)和PCR体系进行检测,比较2种方法的检测结果的准确性。
2 结果与分析
2.1 SD血清型特异性基因筛选
本研究利用BLASTn系统对SD基因组的每个基因的同源性进行分析,获得13个SD血清型准特异性基因,并以此为模板分别设计引物,以实验室保存的多株SD菌基因组为阳性模板,其他非SD菌株基因组为阴性模板进行PCR扩增。结果表明,其中4对引物阳性无条带(结果未展示),另外8对引物中的5对(D1、D2、D3、D11和D12)表现出良好的特异性,只在SD菌液中有扩增条带,在其他非SD血清型中无扩增条带,以上5对引物为SD血清型检测的特异性引物,其模板为SD菌的血清型特异性检测靶基因。剩余3对引物在非SD血清型的沙门氏菌也获得扩增条带,结果见表2。

表2 试验菌株及引物特异性验证结果
续表2

菌株来源数量PCR结果RU61_00441RU61_00445RU61_00447RU61_RS23380RU61_00448RU61_RS09205RU61_RS06985RU61_RS06990invAS. BonnCICC216771---++--++S. CholeraesuisATCC133121---++--++S. ThompsonCICC214801---++--++S. PotsdamCICC215001---++--++S. BraenderupATCC198121---++--++S. BonariensisCICC214961---++--++S. KentuckyCICC214881---++--++S. BazenheidCICC215871---++--++S. ThphiCMCC500711---++--++S. EnteritidisCICC215271---++---+S. EnteritidisCICC214821---++---+S. EnteritidisCVCC33741---++---+S. EnteritidisCMCC500411---++---+S. EnteritidisCMCC500711---++---+S. Enteritidis∗2---++---+S. DublinCICC214971---++---+S. DublinCMCC507611---++---+S. BovismorbificansCICC214991---++---+S. AgonaCICC215861---++---+S. MiamiCICC215091---++---+S. EastbourneCICC215081---++---+S. AnatumCICC214981---++---+S. MleagridisCICC215111---++---+S. London∗1---++---+S. SenftenbergCICC215021---++---+S. AberdeenCICC214921---++---+S. BlockleyCICC214891---++---+S. AdelaideCICC215051--------+S. WandswerthCICC215041--------+Escherichia coliATCC351501---------Escherichia coliATCC438891---------Enterococcus.faecalisATCC129531---------Enterococcus.faecalisATCC292121---------Enterococcus.aviumATCC140251---------Klebsiella pneumoniaeATCC138841---------Staphyloccocus aureus ATCC292131---------Staphyloccocus aureus ATCC259231---------Serratia marcescens CICC101871---------Bacillus pumilusCMCC632021---------Bacillus cereus∗21---------Pseudomonas fluorescens∗21---------Listeria grayiCICC216701---------Listeria seeligeriCICC216711---------Listeria welshimeriCICC216721---------Listeria monocytogenesCICC216621---------Listeria ivanoviiCICC216631---------Listeria innocuaCICC104171---------
注:+: 阳性结果;-: 阴性结果; *:本实验室保存菌株
2.2 多重PCR检测体系特异性评价
如表2所示,当样品中含有SD菌时,多重PCR能同时扩增171、284和512 bp(D1、139-141和D3引物),当样品中含有非SD血清型沙门氏菌时,只能扩增出284 bp;当样品中无沙门氏菌时,无扩增条带产生。此结果表明,该多重PCR反应体系对SD血清型检测具有较好的特异性。
2.3 多重PCR检测体系灵敏度评价
测定SD菌基因组DNA质量浓度为9.580 2 ng/μL,经无菌水按一定比例梯度稀释后进行PCR扩增。如图1所示,当模板质量浓度为383.2 pg/μL时,3对引物均能获得正确的扩增条带,表明该反应体系的模板灵敏度为383.2 pg/μL。

M:DL2000; 1~7:9.580 2 ng/μL, 1.916 ng/μL,383.2 pg/μL, 76.642 pg/μL, 15.328 pg/μL, 3.066 pg/μL和613.13 fg/μL图1 多重PCR检测体系(DNA)灵敏度
Fig.1 Sensitivity of multiple PCR for detection of DNA
将SD菌液的菌落浓度从5.2×104CFU/mL稀释到5.2 CFU/mL,各稀释度菌液取1 mL转接到LB液体培养基中,37 ℃过夜培养,PCR检测。图2结果表明,当菌落浓度为52 CFU/mL时,能同时获得3条清晰的扩增条带,表明此时菌落浓度为该检测特性的菌落灵敏度。

M-DL2000;1-6:ddH2O (对照);5.2×104;5.2×103;5.2×102;52;5.2 CFU/mL图2 多重PCR检测体系(DNA)灵敏度
Fig.2 Sensitivity of multiple PCR for detection of Salmonella spp. and S. Derby
2.4 多重PCR检测体系抗干扰能力评价
将SD菌液和3类样品的背景菌群(不含沙门氏菌),分别按照1∶1、1∶102、1∶104、和1∶106的菌落浓度进行混匀,进行多重PCR扩增。如图3所示,当与3种背景菌群菌落浓度比例为1∶104时,多重PCR均能获得3条清晰的扩增条带,表明在104倍的自然杂菌干扰情况下,仍能获得阳性检测结果,说明该反应体系对环境的自然背景菌群有较好的抗性。
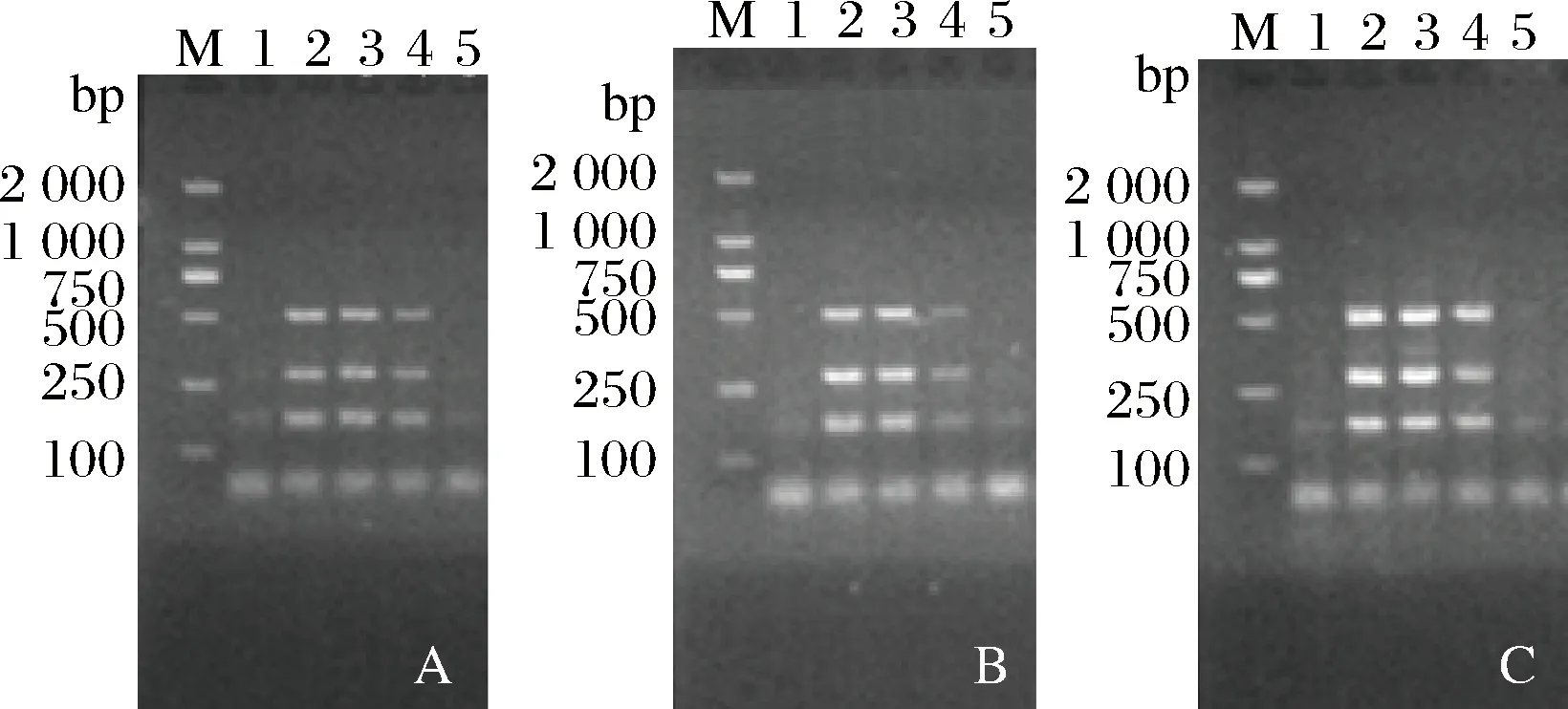
A~C: SD与猪肉背景菌群、鸡肉背景菌群和牛肉背景菌群混合1~5: SD与自然背景菌群混合的浓度比:ddH2O(对照),1∶1, 1∶102, 1∶104, 1∶106图3 自然背景菌群存在对SD检测的影响
Fig.3 Interference tests of multiplex PCR detection of Salmonella spp. and S. Derby with other food background microflora
2.5 多重PCR检测体系人工污染评价
将不同浓度的SD菌液分别污染猪肉、鸡肉和牛肉样品,经37℃培养6、8、10和12 h,多重PCR检测。图4结果表明,当增菌培养6 h后,3种样品的检测限均为3.8×105CFU/25g;当增菌培养8 h后,污染浓度为3.8 CFU/25g的牛肉样品能获得3条清晰的PCR扩增曲线;当增菌培养10 h后,各个浓度污染的3种样品的检出率均为100%。综上所述,当猪肉、鸡肉和牛肉样品增菌10 h,该多重PCR检测体系的检测限能达到3.8 CFU/25g。
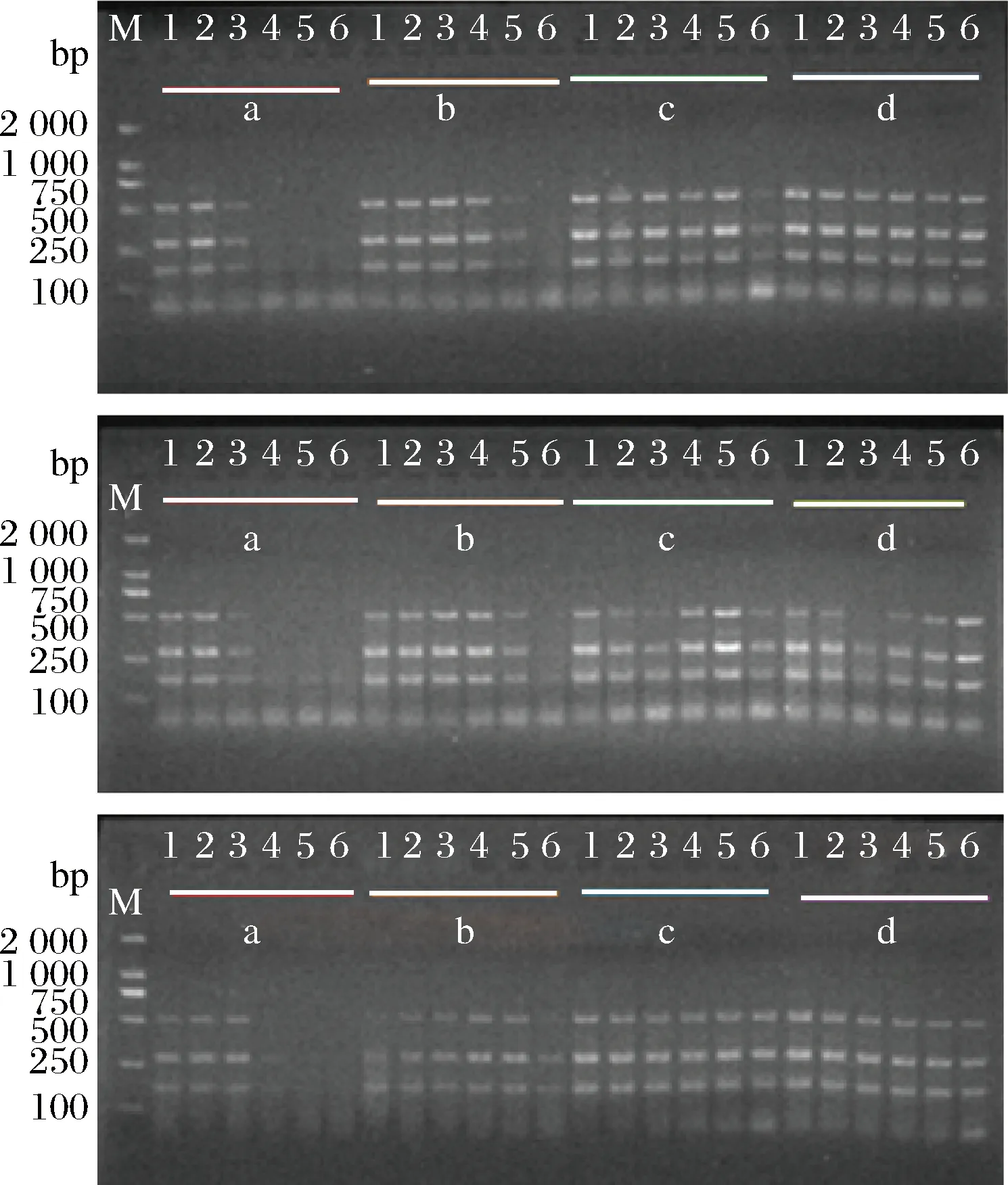
Ⅰ-Ⅲ:人工污染猪肉、鸡肉和牛肉样品;a~d:各样品分别增菌6,8, 10和12 h进行mPCR检测;M: DL2000 marker;1-6: 3.8×105;3.8×104;3.8×103;3.8×102;38;3.8 CFU/25 g图4 SD人工污染猪肉、鸡肉和牛肉样品的检测
Fig.4 SD detection results of different enrichment time for artificially contaminated meat samples
2.6 多重PCR检测体系的准确度评价
将不同浓度的SD菌液分别污染猪肉(21份)、鸡肉(15份)和牛肉(17份),以上3类再各取3份无SD污染的样品作为空白对照,计算该检测方法的灵敏度、特异性和准确度。如表3所示,只有污染6.4 CFU/25g猪肉样品的检测过程中出现了一次偏差,其他2种污染样品的检测结果均与传统培养的检测结果一致,表明该检测方法的准确度达到98.5%。
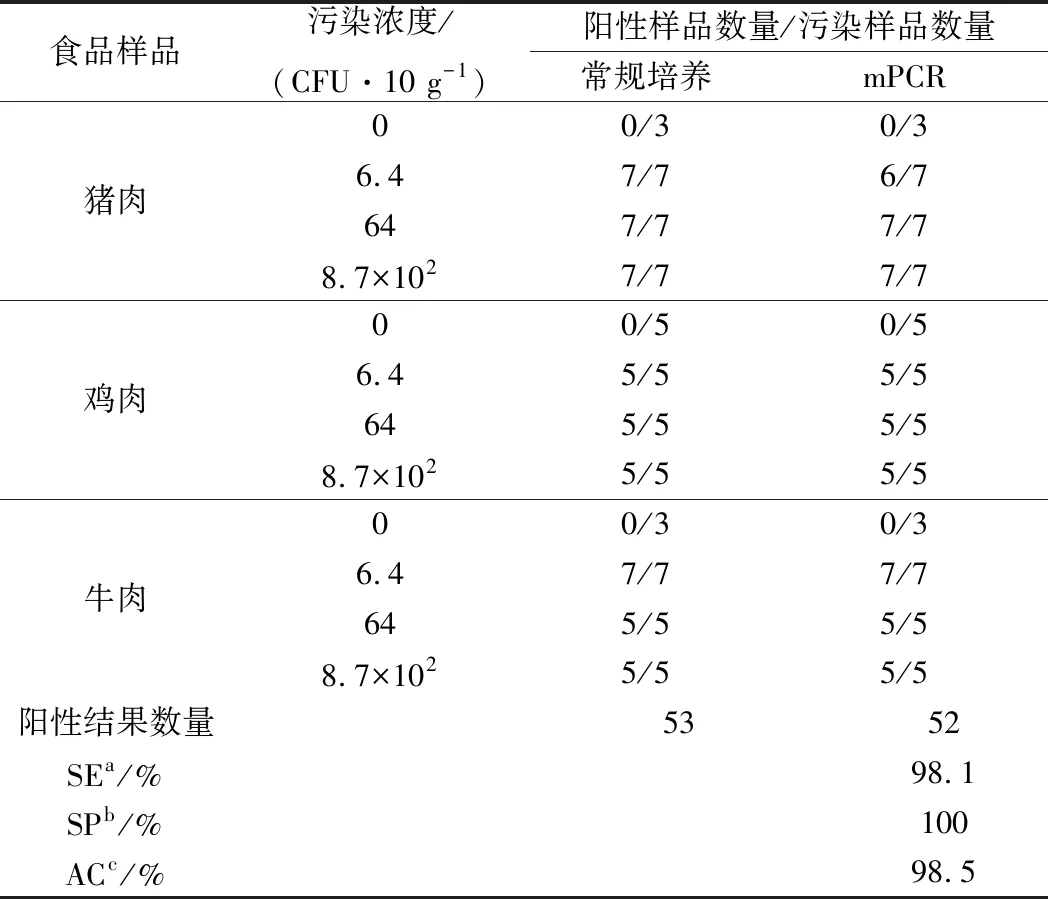
表3 多重PCR检测方法的灵敏度、特异性和准确度
注: SEa,灵敏度;SPb,特异性;ACc,准确度。
3 结论与讨论
沙门氏菌污染具有地域性特点,不同地区的优势血清型存在着一定差异,因此针对沙门氏菌血清型的检测就变得尤为重要[15]。经调查表明,SD血清型主要污染猪肉制品,而且我国是猪肉消费大国,因此对于SD血清型的快速检测技术的开发将变得尤为重要[8,10]。随着分子生物学技术的不断发展, SD血清型的基因草图已经被绘制出来[7]。本研究利用比较基因组技术筛选了SD多个血清型准特异性基因,通过PCR验证最终获得5个该血清型特异性基因。通过分析,选择RU61_00441和RU61_00447基因为沙门氏菌德尔卑血清型的检测靶点,同时结合沙门氏菌检测靶点(invA)共同构建多重PCR检测体系。
传统筛选血清型特异性基因多采用O抗原或H抗原合成基因作为检测靶点,因此需要多轮多重PCR的检测才能确定检测结果[16]。该方法虽然相较于传统培养的方法更加快速和便捷,但由于沙门氏菌血清型种类较多,由于某些血清型的亲缘关系较近,会干扰目标血清型的鉴定,容易造成假阳性或假阴性结果的出现。本研究采用比较基因组的方法进行沙门氏菌血清型的分子检测,同时避免以上的问题,而且通过该方法已经筛选到肠炎沙门氏菌、鼠伤寒沙门氏菌和伤寒沙门氏菌的血清型特异性检测靶点。该检测体系中选用2个德尔卑血清型特异性基因作为检测靶点是为增加检测的准确性,避免假阳性或假阴性结果的干扰。
由于本体系采用多重PCR进行检测造成体系的DNA检测灵敏度高于之前的报道[17]。但经过增菌培养后,活菌的检测灵敏度达到52 CFU/mL,该结果与WOODS等的报道结果相似[12]。在自然环境下,沙门氏菌不是单独存在,生鲜样品中必然含有大量的背景菌群对目标菌群的检测具有干扰作用。该研究按照不同比例将猪肉、鸡肉、牛肉的背景菌群与SD进行混合,利用蒸煮法提取混合DNA进行检测,结果表明,当背景菌群的浓度高于目标菌群104时,仍能获得阳性检测结果,表明该检测体系具有良好的抗干扰能力。同时,本方法采用蒸煮法提取DNA,能有效降低检测成本,提高检测效率[18]。通过人工污染实验表明,该检测经过10 h的增菌培养,在3种食品基质条件下检测限均能达到3.8 CFU/25g。通过对53份样品污染的检测结果表明,该检测体系表现出较好的灵敏性、特异性和有效性。
