缩酮席夫碱类阴离子识别的量子化学研究
2019-04-29梁秋群梁楚欣张淑芬
梁秋群, 刘 峥, 梁楚欣 , 魏 席, 张淑芬,2
(1. 桂林理工大学化学与生物工程学院 电磁化学功能物质广西区重点实验室,桂林 541004; 2. 大连理工大学精细化工重点实验室,大连 116024)
1 引 言
随着经济与科学技术的发展,阴离子在医药、生命科学、环境保护、农业生产和工业催化等领域的研究有着愈来愈重要的影响,因此阴离子识别技术与阴离子受体的设计合成,有着广阔的发展前景[1,2]. 但是高效、高选择性的阴离子受体的合成,目前仍然存在盲目性,这是因为在设计阴离子受体结构时,一方面要考虑与客体分子的结合部位及发射信号部位的设计,另一方面还要考虑受体与客体分子结合过程,其构象的变化,要与客体分子结构相匹配,只有这样才能设计出高选择性、高灵敏度的阴离子识别剂[3,4].
进入新世纪以来,尤其是近十年以来,随着量子化学理论与计算机科技的迅猛发展,整个化学学科都受益良多,并发生着深刻的变化[5-9]. 在超分子化学领域,量子化学的计算结果,可以给出主客体间的相互作用信息,如模拟受体分子与阴离子间的结合方式,计算受体分子与阴离子结合物的构象信息,电荷分布,前线轨道能量和结合能等信息,为新型阴离子受体的设计与合成提供理论指导等[10].
本文利用高斯软件,采用一种常用的密度泛函(Density Functional Theory,DFT)B3LYP,在LANL2DZ水平基组上[11,12],对自制具有缩酮结构的阴离子受体R3-1,R3-2和R3-3及它们与阴离子结合物进行了结构与频率的优化. 从空间结构、电荷分布、结合能等方面,研究了三种受体分子结构性质的差异、阴离子识别过程中受体分子构象的变化以及主客体间的超分子作用.将量子化计算结果与实验结果进行对比分析,进一步探究受体分子对阴离子的识别机理,为新型阴离子受体分子的设计与合成提供量子化理论基础.
2 计算模型与方法
(1)用ChemBio Office2010中的ChemDraw画出三种阴离子受体的分子式,在 Chem3D中打开已经画好的分子式,用MMFF94程序包优化其键长和键角.
(2)将优化好的阴离子受体分子构象保存为gjf格式,导入GaussView5.0,分子构象采用笛卡尔坐标系,采用Gaussian03程序包中的密度泛函理论(DFT)在B3LYP/LANL2DZ基组下进行几何优化和频率分析[12,13],得到在基态下分子稳定的构象、红外光谱、净电荷分布与能量信息.
(3)将计算得到的受体分子与离子稳定构象导入GaussView5.0,采用Gaussian03程序包中的密度泛函理论(DFT)在B3LYP/LANL2DZ基组下,对主客体结合物进行几何优化和频率分析,得到结合物的稳定构象、净电荷分布与能量信息.
3 结果与讨论
3.1 阴离子受体的合成及阴离子识别评价
3.1.1阴离子受体的合成
本文以1,10-邻菲罗啉为起始原料,经浓硫酸/浓硝酸混酸氧化,生成1,10-邻菲罗啉-5,6-二酮;而后在对甲苯磺酸催化下,分别与苯肼、4-硝基苯肼和2,4-二硝基苯肼,反应生成三种缩酮席夫碱阴离子受体R3-1、R3-2和R3-3.
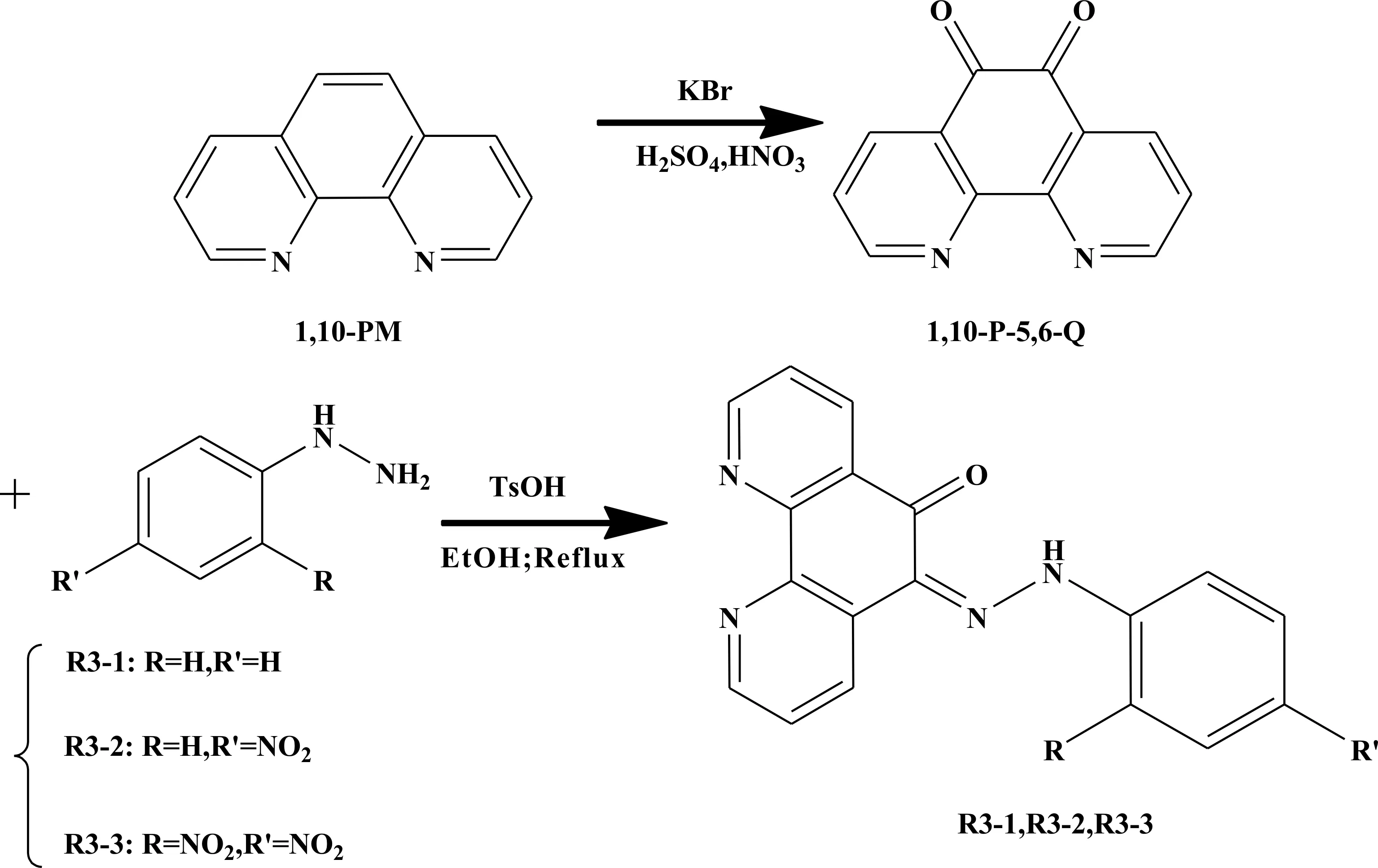
阴离子受体R3-1~R3-3的合成路线Synthetic routes of anions receptors R3-1~R3-3
3.1.2阴离子识别评价
在10 mL浓度为2×10-5mol/L的R3-1或R3-2或R3-3乙腈溶液中,加入100 μL浓度为2×10-2mol/L的各阴离子的四丁基氟铵盐乙腈溶液([A]/[R]=10),测定溶液的紫外-可见吸收光谱,并与三种阴离子受体的吸收光谱图作对比,其中[A]表示阴离子(anion)的浓度;[R]表示受体(receptor)的浓度.实验结果见图1、图2、图3.实验结果表明,在浓度为2×10-5mol/L受体分子的乙腈溶液,加入阴离子前后,溶液吸收光谱中吸收峰的位置与强度发生了变化,可实现对阴离子的识别甚至是定量的分析.三种受体分子中R3-3的识别效果最佳,对阴离子识别灵敏度高低顺序为R3-3 > R3-2 > R3-1.当加入阴离子后,图3显示,受体分子R3-3原480 nm处的吸收峰减弱或者消失,同时在580 nm处有新的吸收峰出现,且该峰的强度随加入阴离子的类别或浓度的不同而改变,加入相同量的各类阴离子对应的吸收峰强度顺序是:F-> AcO-≈ OH-> H2PO4-.
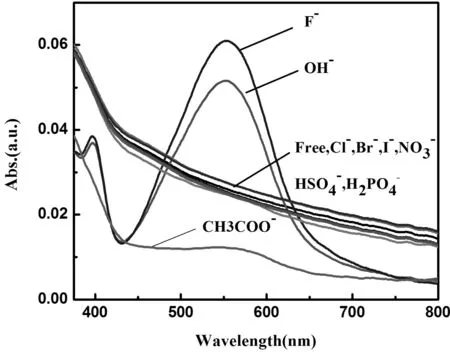
图1 R3-1及加入不同阴离子的紫外-可见吸收光谱([A]/[R]=10)Fig. 1 UV-vis absorption spectra of R3-1 upon addition of anions ([A]/[R]=10)

图2 R3-2及加入不同阴离子的紫外-可见吸收光谱([A]/[R]=10)Fig. 2 UV-vis absorption spectra of R3-2 upon addition of anions([A]/[R]=10)
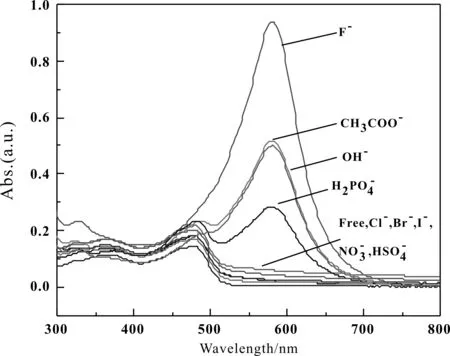
图3 R3-3及加入不同阴离子的紫外-可见吸收光谱([A]/[R]=10)Fig. 3 UV-vis absorption spectra of R3-3 upon addition of anions([A]/[R]=10)
3.2 受体分子的量子化学计算
采用量子化学密度泛函理论,在B3LYP/LANL2DZ基组下对受体分子、阴离子及受体分子和阴离子的结合物可能存在的构象,进行几何优化和频率优化,得到基态下能量最低的稳定构象[14].
3.2.1构象分析
阴离子受体分子的稳定构象如图4所示.

图4 阴离子受体分子的稳定构象Fig. 4 The structures of anion receptors
阴离子受体分子的结构参数见表1.
由图4及表1,我们发现,阴离子受体R3-1、R3-2和R3-3的稳定构象极为相似. 但因受体R3-2和R3-3分子中有吸电子基团-NO2的存在,苯环与邻菲罗啉核的桥链(-NH-N=)发生一定的扭曲,导致R3-2和R3-3分子的共平面性比R3-1较差,如在受体分子R3-1中,二面角∠C7-C8-C9-N16=179.8825°,∠C20-C19-C18-N17=179.9844°,而在R3-2中,这两个二面角分别为179.8079°和179.9755°,再到R3-3中,这两个二面角分别为-179.0493°和175.6782°.

表1 阴离子受体分子的结构参数
受体分子由1,10-邻菲罗啉-5,6-二酮和(硝基)苯肼反应得到,反应新生成的键是C9和N16间的C=N键,研究该键对深入探究这一体系中三个受体性质的关系,有着重要的意义. 在三个受体分子中,C=N键的长度分别为1.3472、1.3397和1.3216 Å,受体分子R3-3的C=N键长最短,该键断裂需要的能量更大,也就最稳定,这是因为R3-3的苯环一端,含两个硝基-NO2取代基,使整个分子的负电荷中心向苯环端偏移,降低了C=N附近的电子密度[15]. 与R3-3相比,R3-2和R3-1的稳定性则要差一些,这与本文3.1.2节得到的结论相互佐证.
硝基的存在,还对亚氨基N-H的键长及强度也有一定的影响,由表1可知,受体分子R3-1、R3-2和R3-3的N-H键长分别为1.4143、1.3403和1.0316 Å,即分子内硝基的存在使N-H键键长变短.较短的N-H键易通过氢键,将阴离子牢牢束缚在受体周围,并将信号传递给受体的信号部[16].
3.2.2红外光谱
经B3LYP/LANL2DZ基组下进行几何和频率优化后,得到的三种阴离子受体分子的红外谱图如图5所示.

图5 阴离子受体计算红外光谱(左)和实测红外光谱(右) Fig. 5 Optimized IR spectra of anion receptors
在阴离子R3-1~R3-3的计算红外光谱图中,均在1450 cm-1和1650 cm-1附近出现强烈的振动峰,分别对应受体分子中的羰基C=O和席夫碱特征官能团C=N双键,且与受体分子的红外光谱基本相似,证明选用B3LYP/LANL2DZ基组用于模拟计算缩酮席夫碱阴离子受体结构参数具有较高的准确性.
3.3 受体分子与阴离子结合物的量子化计算
采用量子化学密度泛函理论,在B3LYP/LANL2DZ基组下对受体分子和F-结合物可能存在的构象,进行几何优化和频率优化,得到基态下能量最低的稳定构象. 阴离子受体分子与F-结合物的稳定构象如图6所示.
阴离子受体分子和F-结合物的结构参数见表2.
由图6及表2,我们发现,阴离子受体分子的阴离子结合位点位于亚氨基-NH-部,受体分子和F-间通过氢键相互作用(N-H…F-),此时受体分子内一些化学键的键长与键角发生改变,分子结构扭曲加剧,共平面性变差. 结合后,受体分子的C=N键分别变长了0.0204、0.0140和0.0200 Å,相互作用后R3-1、R3-2和R3-3的C=N键长分别为1.3676、1.3537 Å和1.3416 Å.
此外,结合物中N-N键与N-C键(苯环)都变短,表明受体分子与阴离子间确实发生了较强的相互作用,削弱了亚氨基N-H的成键能力,N-H键的键长也由原来的1.4143、1.3403和1.0316 Å,分别增长为1.5479、1.5622和1.5804 Å.受体分子R3-1、R3-2和R3-3和F-间形成氢键,氢键的键长依次为1.0234、1.0169和1.0111 Å,即随着苯环端硝基的增加,氢键键长依次变短,主客体间的结合作用依次增强[17].
3.4 净电荷分布
受体分子和结合物中主要原子的静电荷分布见表3.

图6 阴离子受体分子与F-结合物的稳定构象Fig. 6 The structures of anion receptors /F- complexes
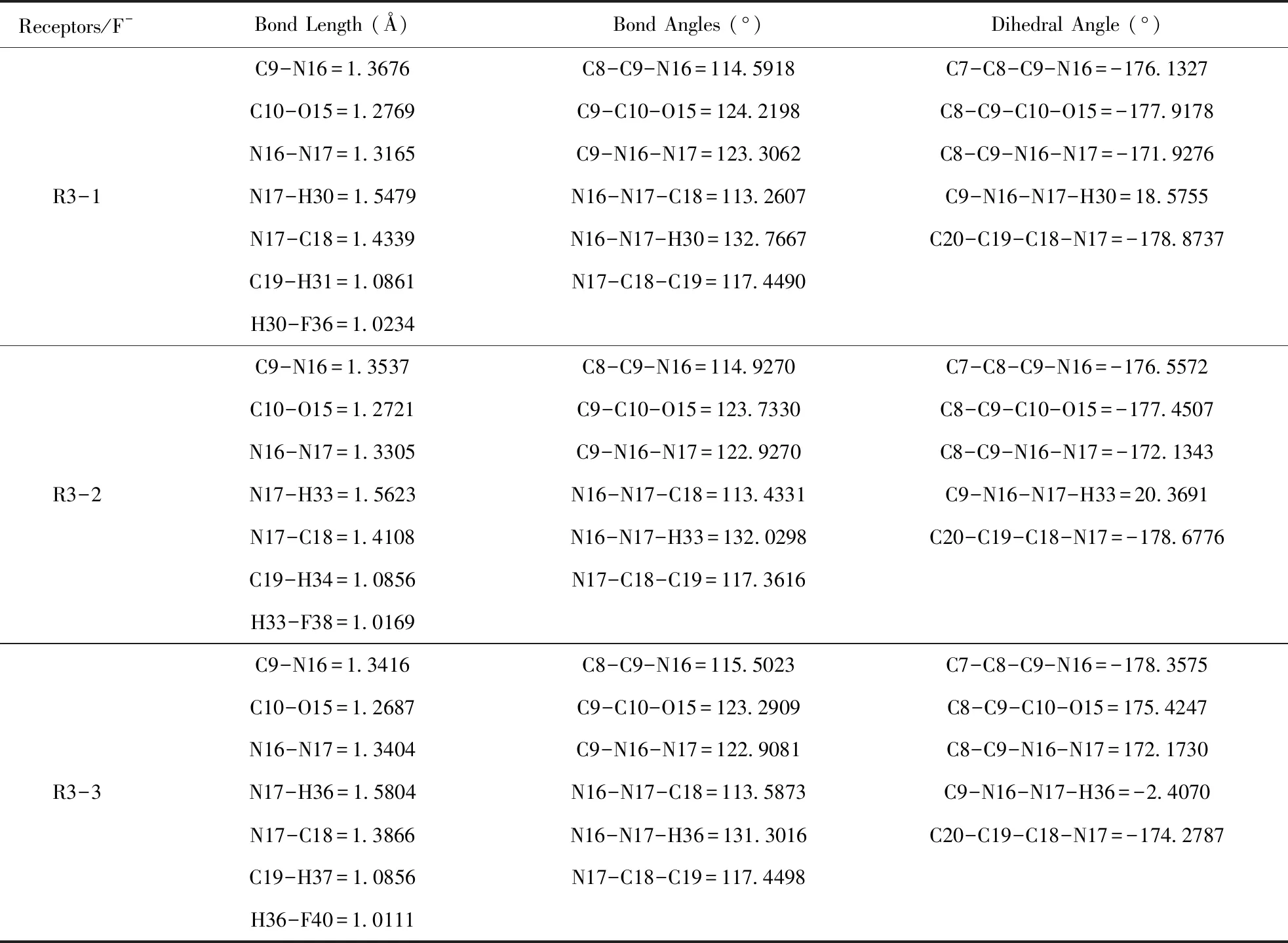
表2 阴离子受体分子与F-结合物的结构参数

表3 受体分子与结合物中原子的静电荷
由表3可以看出,受体分子R3-1、R3-2和R3-3中,席夫碱C=N(C9,N16)的负电荷密度依次减小,C=N键的负电荷密度越小,稳定性越好,电荷分布分析结果与构象分析及实验结果相符. 当F-加入并分别与3种受体分子结合后,F-的净电荷由原来的-1,变为-0.559、-0.550和-0.543,即有一部分负电荷由阴离子转移到受体分子中,且转移的电荷量为R3-3 > R3-2 > R3-1.受体分子中,阴离子的结合位点为亚氨基(NH),由上表可以发现,当受体分子与F-的结合后,亚氨基的负电荷密度明显减小,其中N17由原来的-0.405、-0.409和-0.413变为-0.322、-0.311和-0.290,亚氨基氢N-H(H30、H33和H36)也由原来的0.380、0.385和0.388变为0.503、0.502和0.502,原因是受体分子和F-结合后,亚氨基发生极化,负电荷密度减小,更有利于F-的结合.
另外,在受体分子与阴离子结合物中,邻菲罗啉一侧的桥头碳(C9)负电荷密度较受体分子降低,而(硝基)苯肼一侧的桥头碳(C18)负电荷密度则升高,可能的原因是,F-的加入促进了受体分子中电子向分子的负电荷中心,即(硝基)苯肼部迁移,尤其当苯环上连有吸电子基团时,这种现象更加明显[18].
3.5 能量分析
经量子化学计算,受体分子、阴离子及受体分子与阴离子结合物的能量(单位:Hartree)见表4. 其中,Ecom为受体分子与阴离子结合物的能量,ΔE=Ecom- (ER+EA)为结合物与基态主客体总能量的差值,即受体分子和阴离子结合过程释放的能量,称之为结合能[19,20].

表4 结合物形成过程中的能量变化(Harrtree)
由表4,可以发现一些规律:(Ⅰ)同种阴离子受体分子与阴离子结合能的绝对值大小顺序为:F-> AcO-> H2PO4-,即F-最容易被识别,AcO-次之,H2PO4-再次;(Ⅱ)同种阴离子与受体分子结合能的绝对值大小顺序为:R3-3 > R3-2 > R3-1,即三种受体分子中,R3-3对阴离子的的识别效果最佳,R3-2次之,R3-1再次. 本节结论和构象分析及净电荷分析结论一致,也可以与3.1.2实验结果相互佐证.
4 结 论
本文利用密度泛函理论,在B3LYP/LANL2DZ水平基组下,对本文合成的三种缩酮席夫碱阴离子受体分子R3-1,R3-2和R3-3,以及受体分子和F-的结合物进行量子化学计算,结果表明,F-通过氢键与受体分子的亚氨基相互结合.结合后,受体分子结构扭曲,C=N键拉长. 受体分子R3-1、R3-2和R3-3和F-间形成氢键,氢键的键长依次为1.0234、1.0169和1.0111 Å,即随着苯环端硝基的增加,氢键键长依次变短,主客体间的结合作用依次增强.能量分析表明,考察的三种阴离子中,F-与受体分子的配合能力最强;受体分子中,R3-3对阴离子的识别效果最佳.计算模拟结果,与实验情况基本一致.
