MYO15A与遗传性耳聋
2019-03-04张静关静王秋菊
张静 关静 王秋菊
1解放军总医院耳鼻咽喉头颈外科解放军耳鼻咽喉研究所(北京100853)
2天津医科大学总医院耳鼻喉科(天津300052)
肌球蛋白XVa是肌球蛋白(Myosin)超家族中的一员,MYO15A编码的肌球蛋白XVa除在人内耳内外毛细胞中表达外,在人脑垂体、肝脏、卵巢、睾丸、肾脏等组织亦有表达。MYO15A编码的肌球蛋白XVa可以维持耳蜗毛细胞内肌球蛋白组织结构及毛细胞静纤毛的伸长,对人耳维持正常听觉具有重要意义[1]。因此,人类MYO15A基因突变(NM_016239)可导致常染色体隐性遗传非综合征型耳聋DFNB3(OMIM 600316)(Autosomal recessive forms of non-syndromic deafness 3),并可能与伴有感音神经性耳聋的史密斯•马吉利综合征(Smith-Mage⁃nis syndrome)相关。MYO15A突变最初被发现是源于1995年Friedman等发现印度尼西亚巴厘岛一个神秘的采用独特手语进行交流的村落---Beng⁃kala村,这个村落中2185名村民中有47名(2.2%)患有极重度耳聋,这些耳聋患者符合常染色体隐性遗传非综合征型耳聋特点,对这些患者进行连锁分析后首次定位于常染色体17p11.2一段3厘摩(cen⁃timogan,cM)的范围,命名为DFNB3基因座位,这是继DFNB1(GJB2及GJB6基因突变)及DFNB2(MYO7A基因突变)之后报道的第三个常染色体隐性遗传非综合征型耳聋基因座位。1998年,在3个无亲缘关系的DFNB3家系中证实MYO15A的纯合突变(p.Asn2111Tyr,p.Ile2113Phe及p.Lys2601*)是导致这3个家系致病的原因[2,3]。至此,开启了对MYO15A基因突变的全面深入研究,内容涵盖MYO15A基因结构和功能、利用鼠模型对MYO15A突变致病机制研究、基因型与表型相关性、新一代测序技术应用于MYO15A突变检测以及MYO15A基因治疗等领域,本文总结了二十年来与MYO15A突变相关的遗传性耳聋研究。
1 MYO15A是导致常染色体隐性遗传非综合征型耳聋一个值得关注的常见基因之一
自从1995年印度尼西亚巴厘岛Bengkala村MYO15A基因突变导致常染色体隐性遗传非综合征型耳聋(autosomal recessive non-syndromic hear⁃ing loss,ARNSHL)DFNB3被首次报道,迄今已有包括阿尔及利亚、摩洛哥、印度、巴基斯坦、巴勒斯坦、土耳其、伊朗、卡塔尔、阿曼、突尼斯、以色列、中国、韩国、日本、捷克、德国、法国、美国、墨西哥、巴西、荷兰、西班牙等20余个国家、地区报道了200余个MYO15A突变位点[4-51]。由于中东及南亚等国家地区在人口学存在近亲婚配及大家族更为普遍的特点,该地区MYO15A突变引起ARNSHL的研究报道更为多见,并且相比于其他国家及地区,这些地区MYO15A基因突变在ARNSHL中所占的比例也较高[6,9,15-18,22,23,27,52,53]。9.9%的土耳其ARNSHL家系与MYO15A突变相关,在所有引起ARNSHL的基因突变中位列第二位[16]。Liburd等在2001年巴基斯坦112个ARNSHL家系中,连锁到DFNB3基因座位的家系占10%(11/112),最终证实为MYO15A基因突变的家系占5%[52]。随后,巴基斯坦一项研究中,MYO15A突变占ARNSHL比例为3.33%(20/600)[6]。两项土耳其遗传性耳聋的研究报道表明,MYO15A突变导致ARNSHL的比例分别为3%及6.2%[17,18]。Fattahi等报道在伊朗5.71%的ARNSHL与MYO15A突变相关,在该国所有引起ARNSHL的基因中位列第二位[22]。在东亚地区,ARNSHL中MYO15A突变比例相对较低,韩国为2.1%,排在SLC26A4、GJB2及 CDH23之后位列第四位,日本这一比例为1.67%(10/600)[54]。中国汉族的一项研究表明190例非综合征型耳聋患者中,有5例为MYO15A突变[55],另外在中国少数民族维吾尔族人群中先后报道了4个MYO15A突变导致遗传性耳聋的家系[31,56]。
2 MYO家族及MYO15A的结构
肌球蛋白超家族是一组能够与细胞骨架肌动蛋白丝结合,水解ATP产生能量,并使其产生运动的分子马达超家族。肌球蛋白对于维持细胞的形态、细胞的运动、细胞器的转运及细胞信号的传导等具有重要的生理作用。通常肌球蛋白结构分为头部、颈部及尾部结构域。头部(N-末端)结构保守,可以结合肌动蛋白丝水解ATP获能;颈部包括一个或多个IQ机构域,每个IQ提供一个与钙调蛋白或特异性肌球蛋白轻链结合的位点;(C-末端)为多变结构域,赋予各类肌球蛋白独特功能尾部,如二聚化、与膜结合或者成为膜蛋白结合的靶点。肌球蛋白超家族中编码的肌球蛋白对于维持正常的听觉功能有重要的作用,编码上述肌球蛋白的基因突变会造成不同类型的遗传性耳聋:MYO1A(DF⁃NA48)、MYO3A(DFNB30)、MYO6(DFNA22/DFNB37)、MYO7A(DFNA11/DFNB2)、MYO15A(DFNB3)、MYH9(DFNA17)、MYH14(DFNA4)。肌球蛋白根据有无双极性极丝可分为传统型肌球蛋白(conventional my⁃osin)及非传统型肌球蛋白(unconventional myo⁃sin)。常规肌球蛋白主要为分布于肌肉细胞的MYO2,而肌球蛋白XVa属于非常规肌球蛋白,位于耳蜗及前庭毛细胞静纤毛的顶端[1]。MYO15A基因包含66个外显子,长71,097 bp,它转录的mRNA最长达11,876 nt,可编码多种剪接转录本[2]。MYO15A基因编码最长的转录本包括35390个氨基酸残基(分子量为365 KDa),其中2号外显子编码一段延伸的N-末端,其后是头部的马达(Motor)结构域,包括:ATP及肌球蛋白的连接位点、I型及II型螺旋转换器、后续螺旋、SH3螺旋、SH1/SH2螺旋及一个转换器结构域。根据其头部有无由MYO15A基因2号外显子编码的一段N-末端区域,可将编码的肌球蛋白XVa分为两个亚型,肌球蛋白XVa-亚型1存在由2号外显子编码的N-末端区域,肌球蛋白XVa-亚型2则无N-末端区域。颈部结构域由两条轻链组成(IQ基序)。最长的尾端结构域是由2个肌球蛋白尾部同源物4(myosin tail homology 4,MyTH4)、2个 FERM结构域(F,ezrin,radixin,and moesin)、1个SH3结构域(src homology 3)及1个C-末端I型PDZ结合基序组成(图1)。
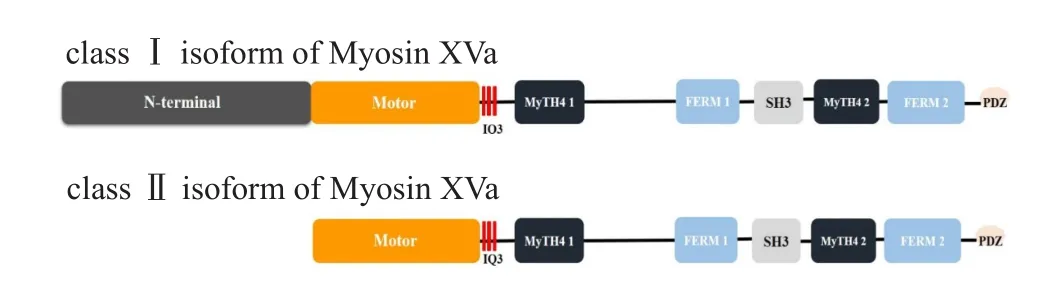
图1 肌球蛋白XVa的两种亚型结构图Fig.1 Structure of two isoforms of Myosin XVa
3 鼠模型对MYO15A突变引发内耳毛细胞形态及功能的研究
Shaker2(sh2)鼠最早报道于1928年,是受到X线电离辐射的鼠的子代,表现为对声音反应差、出现异常摇头及旋转行为。sh2鼠毛细胞肌球蛋白结构异常,毛细胞静纤毛变短。sh2鼠Myo15a位于11号染色体,与人类位于17pl 1.2的DFNB3同源[57]。在纯合突变的sh2鼠的受精卵中注入包含野生型MYO15A的细菌人工染色体BAC425p24(140-kb t)(bacterial artificial chromosomes,BACs),sh2鼠的听力到了恢复,进行繁殖后的后代种系均携带BAC425p2,并且听力正常[58]。正是通过BAC可恢复sh2鼠的听力,编码非常规肌球蛋白的MYO15A最终被鉴别出来。毛细胞纤毛位于耳蜗感觉毛细胞的顶部,在人类及鼠耳中负责机械-电信号的转换。毛细胞纤毛向细胞外突起的结构称为静纤毛,每根静纤毛包括一个肌球蛋白核心,其长度按照排列次序依次增长,形成阶梯状的结构排列[59]。细胞外的侧边突起即侧连接(side-links)可使毛细胞的静纤毛聚集在一起,静纤毛的顶端的连接(tip-links)有助于信号的传递。肌球蛋白XVa位于静纤毛的顶端,在毛细胞纤毛的成熟过程,肌球蛋白XVa的数量与静纤毛的长度成正相关。纯合的sh2鼠由于肌球蛋白马达结构域提前终止了编码造成了肌球蛋白XVa的缺乏,表现为极重度的听力损失,这与人类DFNB3病人的症状极为相似,不过人类无前庭功能低下的表型。与野生型鼠相比,可以明显观察到sh2鼠毛细胞静纤毛在发育过程中分布位置并没有出现异常,但是静纤毛的长度变短,特征性的阶梯状结构消失,而静纤毛的长度及纤毛束的阶梯状结构对于保持正常的听觉功能十分重要[22]。
Whirler(whi)鼠是一种携带突变的whirlin基因的鼠模型,表现为与sh2鼠相似的极重度耳聋表型[60]。纯合突变的whi鼠缺乏whirlin蛋白,这种鼠的静纤毛的形态与sh2鼠十分相近:表现为长度变短及阶梯结构消失。研究表明肌球蛋白XVa与whirl⁃in蛋白互为为伴体(partners),肌球蛋白XVa的PDZ配体与whirlin蛋白的2个PDZ结合区结合,保持两者间的相互作用。肌球蛋白XVa是哺乳动物中已知的唯一拥有PDZ结构域的肌球蛋白。纯合突变的whi鼠表静纤毛的顶端存在肌球蛋白XVa,而纯合突变的sh2鼠在静纤毛的顶端既没有whirlin蛋白,又没有肌球蛋白XVa。肌球蛋白XVa作为马达蛋白具有运动的能力,能够沿肌球蛋白肌丝迁移[61]。这些研究数据都支持肌球蛋白XVa在耳蜗毛细胞纤毛形成阶段扮演转运蛋白的角色对whirl⁃in进行转运的假设,这种转运最可能发生在静纤毛阶梯结构形成及静纤毛变长的阶段。
4 MYO15A突变基因型与听力表型相关性研究
MYO15A突变的基因型与听力表型相关性研究是“精准医疗”时代对遗传性耳聋个体进行精准诊断、评估、预测、治疗和预防的关键,亦是MYO15A突变研究的聚焦点。近二十年来国内外学者对MYO15A突变的基因型与听力表型相关性的认知是逐渐进展的过程。最初十年报道的MYO15A突变导致常染色体隐性遗传性耳聋的听力表型均为先天性双侧全频重度、极重度感音神经性耳聋[52,57,62]。直到2007年,Nal等2007年首次报道了MYO15A的2号外显子编码的N’末端的突变(p.G1112fsX1124)造成低频有残余听力的轻型的听力表型。当时研究认为MYO15A突变患者听力损失表型与基因突变所位于的区域是密切相关的。前述中提及肌球蛋白XVa分为两个亚型,亚型1存在由2号外显子编码的N-末端区域,亚型2则无N-末端区域。研究表明N-末端区域发生突变会导致肌球蛋白亚型1异常,听力表型为有残余听力的非重度极重度感音神经性聋,例如既往报道的MYO15A(p.Arg1112ValfsX1124)[6],MYO15A(p.Tyr289X)[17],MYO15A(p.Glu396Argfs*36)[9]及MYO15A(p.Ser1176Valfs*14)[29]等发生在N-末端区域突变的听力表型为低频区有残余听力的感音神经性聋;而亚型1及2均有突变,即非N-末端区 域(Motor、MyTH4 1、FERM1、SH3、MyTH4 2、FERM2及PDZ结构域)发生的突变,听力表型多为先天性或语前性全频重度-极重度感音神经性耳聋,例如发生在Motor结构域的突变p.Cys1666X[27],MyTH4 1 结 构 域 的 突 变 p.Ile2113Phe、p.Asn2111Tyr[2],FERM1结构域的突变 p.Lys2601*[2,11],SH3结构域的突变p.Val2940fsX3034,MyTH4 2结构域的突变p.Leu3160Phe[6],FERM2结构域的突变p.Asp3320ThrfsX2及PDZ结构域的突变p.Ser3525AlafsX29[45]均表现为先天性重度极重度感音神经性耳聋。Belyantseva等[1]进行的一项Sh2鼠的研究也表明非重度感音神经性听力下降可能与突变发生在2号染色体编码的N-末端延伸结构域有关系。研究者转染一个没有N-末端延伸结构域的肌球蛋白XVa-亚型2进入有异常短小静纤毛Sh2鼠,结果显示静纤毛出现延长,并表现野生型鼠毛细胞纤毛正常的阶梯结构。既往研究表明这种阶梯状结构的异常是导致听力损失的重要的原因,而没有N-末端延伸结构域的肌球蛋白XVa-亚型2就可以进行修复这种静纤毛的异常结构,从某种程度上说明2号外显子编码的N-末端延伸结构域的异常,仅引起功能的异常或轻微的结构异常,表现为较为轻型的感音神经性耳聋[6]。这种MYO15A 2号外显子编码的N-末端区域发生突变与非重度听力表型相关,而发生在其他区域内的突变与重度听力表型相关的研究结果曾经给MYO15A突变的遗传性聋患者的精准医疗提供了分子遗传学依据。根据MYO15A突变位于的不同区域可以预知听力损失的程度,对于突变位于2号外显子编码的N-末端结构域,需要考虑低频残余听力,在人工耳蜗植入术中尽可能保留低频残余听力,并在术后将声电联合刺激(electroacoustic stim⁃ulation,EAS)作为其最佳的听力康复选择;而编码其它结构域的MYO15A突变则需要尽早考虑进行人工耳蜗植入术及言语康复训练。
然而,随后的研究表明MYO15A突变的基因型与听力表型相关性似乎比之前预想的更为复杂。报道了更多的非先天性双耳重极重度感音神经性耳聋的听力表型,表型特征更为多样化:除了低频区残余听力[22],还包括听力曲线为下降型的先天性中重度感音神经性聋[6,17,29,63]、全频中重度感音神经性聋[9]、进展性高频下降型重度感音神经性聋等[32]、迟发性、进展性中重度感音神经性聋(发病年龄最迟达14岁)[34,64]。有趣的是,研究发现在同一MYO15A突变的常染色体隐性遗传性耳聋的家系中,携带相同致病突变的患者也会表现为不同程度的听力表型[17,23]。Cengiz等[17]报道了一个MYO15A的2号外显子发生纯合突变(p.Tyr289*)的土耳其ARNSHL家系,家系成员中包括先证者在内的三个兄妹均表现为先天性重度极重度感音神经性耳聋,而先证者母亲的听力表型为进展性非重度感音神经性聋。另外,随着MYO15A突变报道的日益增多,发现在MYO15A非编码N-末端区域的突变,均有非重度的听力表型的报道(表1)。例如,Chang等报道了两个听力表型为双侧对称性并在低频区存在明显的残余听力的语后聋家系,两个家系的MYO15A突变均为复合杂合突变(p.Arg1835His,p.Ser3417del)及(p.Gln3264*,p.Ile3421Met),突变分别位于Motor区及FERM2区[34]。产生这种非重度的听力表型,可能与如下因素有关:MYO15A等位基因致病性较弱、存在修饰基因减轻了听力损失的程度,环境因素的影响。另外,近年来基因诊断技术的进展也进一步丰富了MYO15A的表型谱。既往对于近亲婚配遗传性耳聋家系的研究常采用连锁分析,那些由纯合突变引起的具有严重听力表型的患者(多来自中东、南亚等近亲婚配习俗的国家及地区)总是优先地纳入到相关的遗传学研究中。然而,随着下一代测序技术的不断发展,可以越来越多地对全世界范围内非近亲婚配的中小家系患者进行分子遗传学诊断,鉴别出更多的不同程度听力损失表型的复合杂合突变患者,使得MYO15A突变表现出更为多样的表型特征。未来需要收集更多MYO15A突变耳聋患者,丰富不同种族地区的MYO15A突变及听力表型数据库,进一步研究MYO15A突变的基因型与听力表型相关性。

表1 目前已报道的非重度听力表型的MYO15A突变Table 1 Overview of all known cases of MYO15Avariations with milder hearing loss phenotype
5 MYO15A基因杂合突变与史密斯-马吉利综合征
史密斯•马吉利综合征(Smith–Magenis syn⁃drome,SMS)临床表现为中度智力障碍及多种先天性异常,包括轻度颅面及骨骼发育异常、行为问题、睡眠障碍、身材矮小、心脏及肾脏发育异常[65],发病率为1/25,000。Greenberg等报道了一组SMS患者,68%(17/25)的SMS患者伴有不同程度及性质的听力损失,其中由反复发作的中耳炎所导致的传导性耳聋患者最多见,感音神经性耳聋及混合性耳聋亦有报道[66]。SMS是由于重复基因簇发生同源重组时导致常染色体17p11.2出现4 Mb的缺失,而DF⁃NB3基因座位也恰巧位于这段缺失的区域[57]。因此,伴有感音神经性耳聋的SMS患者是否存在MYO15A突变成为研究热点。Liburd等[52]对8位伴有感音神经性耳聋的SMS患者进行MYO15A 66个外显子测序,结果检测出一例伴有中重度感音神经性听力损失(高频下降型)的SMS患者MYO15A的31号外显子出现杂合错义突变:p.Thr2205Ile(c.6952 C>T)。通常MYO15A单杂合突变不会导致耳聋,而部分MYO15A单杂合突变的SMS也会出现感音神经性耳聋,推测导致耳聋的可能机制是:存在17p11.2缺失的那条染色体的DFNB3缺失,与此同时另一条完整的17号染色体DFNB3出现了亚效等位基因突变[11]。
6 小结
从1995年首次报道DFNB3家系,并经连锁分析鉴定出MYO15A为DFNB3致病基因的20多年来,随着下一代测序技术的广泛应用,特别是全外显子测序技术的应用,MYO15A突变的报道不再局限于在中东地区等近亲婚配多见的国家,世界范围内MYO15A突变位点的报道日益增多,特别是复合杂合突变位点不断丰富MYO15A基因突变数据库,听力表型呈现出多样的特点:听力损失程度从重度极重度到轻中度感音神经性耳聋,除先天性聋外,语后、进展性感音神经性聋也有报道。MYO15A基因型与表型具有一定的相关性,但研究表明修饰基因、环境因素也可能对听力表型产生影响,这也是今后研究的热点方向之一。另外,近来对MYO15A突变的诱导多能干细胞(induced pluripo⁃tent stem cells,iPSCs)的细胞系进行基因校正与内耳毛细胞的诱导分化取得成功,证明了基因校正能有效逆转MYO15A突变导致的毛细胞样细胞的形态和功能缺陷,这为今后人类MYO15A突变所致感音神经性耳聋的治疗方面带来了希望。
