EYA4突变致DFNA10型遗传性耳聋的研究进展
2019-03-04吴楷文关静王秋菊
吴楷文 关静 王秋菊
解放军总医院耳鼻咽喉头颈外科解放军耳鼻咽喉研究所(北京100853)
EYA4(eye absent 4)是eya家族蛋白的一员,于 1999年被克隆并定位在6q23.2[1],作为转录激活因子参与机体内多种生物活动,对维持Corti器的发育成熟具有重要作用。EYA4的突变与常染色体显性遗传性耳聋(Autosomal Dominant Non-syndrome Hearing Loss,ADNSHL)、扩张型心肌病(Dilated cardiomyopathy,DCM)及多种恶性肿瘤的发病相关[2],其中EYA4突变导致的ADNSHL定位于DFNA10基因座,在世界范围内被报道,其临床特点表现为双侧对称、迟发性、渐进性加重的感音神经性耳聋,可能与单倍体剂量不足导致的失能效应相关。本文旨在将EYA4突变导致的DFNA10型遗传性耳聋的研究进展进行综述,绘制致病突变图谱,分析临床表型与基因型的关联及可能的致聋机制。
1 EYA4的克隆、定位与表达
1996年,O’Neill等对一个美国的ADNSHL家系进行了连锁分析,发现了一个新的耳聋基因座DFNA10,并将其定位在6q22-23一段15cM的范围内,标记D6S474-D6S270之间[3],随后通过扩大家系验证逐渐将范围缩小至6cM,标记在D6S413-D6S292 之间[4]。1999年Borsani等对人类EYA4和小鼠Eya4进行了克隆与定位,并详细描述其特点,EYA4在人类6号染色体恰位于DFNA10区域,小鼠Eya4位于10号染色体,两者的核苷酸编码区序列更88.9%同源,氨基酸序列同源性达到90.9%。由于不同剪切方式导致EYA4转录有多种亚型,含有19外显子的亚型在发育中的内耳表达,含有20外显子的亚型在成熟内耳表达[1]。2001年Wayne等通过单链构象多态性(Single-Strand Con⁃formation Polymor-phism,SSCP)的方法在2个之前报道的DFNA10家系中分析并发现EYA4的两种致病突变,由此确定了EYA4即为DFNA10型耳聋的责任基因[5]。
Eya4在小鼠胚胎期听囊表达,发育过程中,逐渐集中在血管纹及前庭膜表达,由蜗管上层转移到蜗管底部神经上皮表达,出生后则在螺旋神经节边缘、Corti器及骨蜗管表达[5]。eya4在斑马鱼受精24小时后胚胎的腹侧听囊表达,72小时集中在胚胎三处嵴突和两处斑块的感觉区域(内含支持细胞及毛细胞)表达eya4吗啡啉突变体的斑马鱼胚胎表现为体积缩小、听泡发育畸形、毛细胞形态发育异常且数量较野生型减少、听觉功能异常[6]。EYA4在非人类的灵长类动物---狨猴的内耳主要表达在内、外毛细胞,支持细胞和螺旋神经节细胞分布。EYA4在成年狨猴内耳所有支持细胞均有表达,而在啮齿类动物出生后就不再于支持细胞表达[7]。
2 EYA4蛋白功能域及功能
EYA4位于6q23.2,含21个外显子,从2号外显子开始编码,共640个氨基酸,含有2个结构功能域:eya可变区域(eya variable region,eyaVR)和eya同源区域(eya homologous region,eyaHR),两个结构域各行使不同的功能。eyaVR位于N端,其氨基酸序列在不同物种间变化大,保守性差,内含一段相对保守的脯氨酸-丝氨酸-苏氨酸(proline–serine–threonine,PST)丰富(34%–41%)的反式激活区域,行使转录激活功能和苏氨酸磷酸酶活性作用。PST结构对于转录激活是必须的,其通过与SIX/SO(Sine oculis)、DAC/DACH(Dachshund)的协同作用行使转录激活功能,若结构域缺失将严重影响Eya蛋白的正常功能[8-12]。PST结构的苏氨酸磷酸酶活性可通过激活特定信号通路保护机体免受细胞凋亡产生的有害物质,增强机体固有免疫[13]。
eyaHR位于C端,由271个氨基酸组成,在Eya蛋白家族及不同物种间均高度保守[2,14]。目前已知eyaHR有两方面功能:一方面,它能够与SIX/SO和DAC/DACH蛋白相互结合,eyaHR与SIX家族蛋白的相互作用形成共转录因子进入细胞核,SIX蛋白的同源异型结构域与DNA序列结合,Eya蛋白行使激活功能,Eya蛋白与DACH蛋白的相互作用需要CBP蛋白来介导[5,15]。另一方面,eyaHR同时具有酪氨酸磷酸酶活性,而且属于真核动物中少见的卤酸脱卤酶,Eya蛋白通过天门冬氨酸作为亲核体并且结合Mg2+来催化酪氨酸磷酸盐的水解作用,参与多种生物化学反应,如协助癌细胞转移[16]、促进乳腺癌细胞增值并抑制凋亡[17],抑制酪氨酸磷酸酶活性可干扰血管生成[18],此外酪氨酸磷酸化酶活性还具有调解DNA损伤修复的功能[19,20]。
3 EYA4突变与DFNA10型遗传性耳聋
3.1 DFNA10型耳聋临床表型特点
EYA4致病突变导致的DFNA10型耳聋在遗传学方面具有遗传稳定性和重复性,目前在美国等十一个国家共报道19个DFNA10型耳聋家系[5,21-37],不同家系的临床表型既有相似性又有差异性,相似性表现在语后发病、迟发性、进行性加重、双耳对称的感音神经性听力损失,部分家系可伴有耳鸣症状,但无前庭功能异常。差异主要表现在:(1)发病年龄变化差异大。瑞典家系中有患者4岁即发病[30],而美国家系患者中最晚发病年龄为50岁[5],来自中国的6个EYA4家系共60名患者平均发病年龄为30岁,个别发病早至8岁[28,31-33,36];(2)发病初期听力受损特点不同。几乎所有DFNA10患者发病时都累及中频听力,同时部分家系高频或低频可受累,相当一部分家系发病初即全频受累,因此发病初听力曲线常见为U型,也可见下降型、平坦型曲线;(3)听力损失的进展速度不同,比利时家系患者听力下降平均1.08dB/年,55岁之前为轻度听力损失,55岁之后为中-重度[38];美国家系患者在听阈不超过50dB时听力下降约0.6dB/年[39];6个中国DFNA10型家系耳聋患者听力损失为1.3dB-3.9dB/年,在听阈<55dBHL时,平均下降3.8dB/年,听阈>56dBHL时,平均下降2.2dB/年,听力损失在5年病程内为轻度,5-30年左右为中-重度,达30年后(平均达57岁)多进展为重度乃至极重度(图1)[28,31-33,36,40]。所有的DF⁃NA10型耳聋患者的听力损失最终将累及全部频率,成为重度-极重度的平坦型听力曲线。
Frykholm等对瑞典家系耳聋患者的PTA与性别、年龄、双亲来源等因素进行分析,发现男性患者在年轻时即进入中重度听力损失,男性患者的后代耳聋的发病时间更早[30]。van Beelen对来自荷兰的家系进行多年随访分析,发现不同频率听力损失进展不同,2kHz每年下降0.5dB,8kHz每年下降1.6dB,高频进展较快,这与美国家系的特点相似[34]。Choi等在统计韩国中度听力下降的ADNSHL家系中,发现EYA4突变导致的DFNA10耳聋所占的频率为7.14%(1/14)[35],结合另一项研究得出该数据为8.57%(3/35)[25,26]。Kim等在87名ADNSHL患者中检测出2个EYA4致病突变[29],因此在显性遗传性耳聋家系研究中,不能忽略EYA4的致病的可能。
EYA4突变还可引起综合征型听力损失,Sch⁃onberger等人报道了2个无血缘关系的美国家系,其患者同时出现迟发性渐进性听力损失和DCM,20岁出现轻度听力损失并逐渐变为重度-极重度,心脏症状最早31岁出现,通过检查示心脏扩张,NYHA评估心力衰竭为I级-II级,随年龄逐渐加重,40多岁演变为DCM,50-60岁病情加重乃至死亡,必须行心脏移植手术,这是报道的唯一的EYA4突变导致的综合征型听力损失[21]。
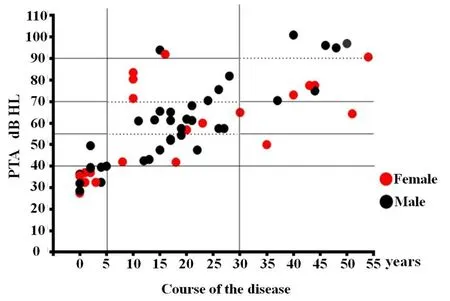
图1 中国DFNA10耳聋患者PTA与病程的分布特点Fig.1 Analysis of pure-tone thresholds and disease course of DFNA10 patients in China共统计60名患者,男性38名,女性22名,轻度听力损失15人,中度-重度听力损失38人,极重度听力损失7人。病程5年内听力损失多为轻度,5-30年为中-重度度,>30年多为重度-极重度A total of 60 patients were involved in this study,including 38 males and 22 females,15 patients with mild hearing loss,38 with moderate to severe hearing loss,and 7 with profound hearing loss.Most DFNA10 patients show mild hearing loss within 5 years of onset,moderate to severe within 5 to 30 years,and severe to extremely severe within 30 years.
3.2 临床表型与基因型关联分析
截止目前共报道EYA4的18种突变会导致DF⁃NA10型耳聋,包括4种无义突变,7种插入(缺失/重复)突变,2种剪切突变,1种大片段缺失突变(Copy number variation,CNV)和4种错义突变(图2)。前四种突变均导致氨基酸翻译提前终止而形成截短蛋白,严重影响EYA4蛋白的功能。采用ACMG指南标准对4种错义突变进行致病性分析,p.G171R、p.I434K、p.T548R突变均为“可能致病的”[28,31,33,40],而p.F326L为“意义不明确的”,其致病性有待进一步明确[41]。
EYA4突变导致的DFNA10型耳聋具有高度异质性,表现在相同或相似的突变可引起不同的临床表型,而相似的临床表型可由不同的突变导致。例如在荷兰家系(致病突变为c.464delC)中,发病初期V-8,V-7,IV-13仅中频轻度受累,而IV-24/25累及中低频,呈中度听力损失;IV-24/25全部频率均随年龄显著下降,III-3/7/14、V-8患者在个别频率下降,III-5/13/16在10年中无明显下降[34]。同样在中国家系(致病突变为c.544-545in⁃sA)中,两不同分支临床特点不同,II-1分支高频受影响呈下降型曲线,II-6分支全频受累呈平坦型曲线[32]。相同或相似的突变导致的不同临床表型,可能与环境因素、个体因素或其他基因修饰有关。相反的,中国家系(致病突变为c.1301T>A)与比利时家系(致病突变为c.1759C>T)相比,突变不同但临床表型相似,发病年龄最早在1-10岁,最晚在40岁之前,首先中频听力受损,为“Cook⁃ie-bite”样听力曲线,病程进展缓慢,约1dB/年,最终听力转归为平坦型曲线[5,28]。
所有的截短突变都影响了eyaHR区域,故一般认为该区域的完整性对于维持听觉功能很重要,但随着越来越多错义突变的报道,发现仅影响eyaVR区域的突变也会引起DFNA10型耳聋,如c.511G>C突变引起单个氨基酸替换,影响eyaVR而与eyaHR无关联,说明eyaVR区域的特殊突变亦影响了听觉功能[31]。c.863C>A(p.S288X)在韩国家系多次报道,是始祖效应突变,在其他不同人群和地区其始祖效应仍需有待验证[25,29]。
EYA4的CNV为涵盖9号外显子、内含子和10号外显子的2238 bp的大片段缺失,导致194位氨基酸处往后移码30个氨基酸,在222位置氨基酸翻译终止(p.D194Gfs*30),该突变导致渐进性听力损失和DCM,Schonberger等人认为EYA4蛋白的193-343区域可能在维持心脏功能中发挥关键作用。2009年报道一名波兰患者的6q23.2-6q24.1之间出现7.7-9.4Mb的片段缺失,波及到EYA4的大部分区域,只剩3个外显子,导致临床表现为小头畸形、身材矮小、动脉导管未闭,感音神经性耳聋、精神发育迟滞、行为异常等,从一定意义上支持了EYA4特定区域的变异会影响心脏和内耳功能的观点[42]。eya4吗啡突变体斑马鱼表现为心腔扩张,肌纤维发育不良,30%左右胚胎围心腔出血心肌射血功能减弱,肌纤维分化不明显,说明了eya4的下调影响心脏发育及功能[6]。但随着临床上更多的研究发现影响EYA4蛋白的193-343区域的缺失突变,如p.D194Yfs*52、p.S288X,甚至更严重的截短突变如p.P155Qfs*43,都仅仅引起单纯的DFNA10型耳聋,无任何DCM症状及猝死的发生[25,29,30,34]。EYA4突变导致合并DCM的综合征型耳聋的机制仍需进一步探索。

图2 与DFNA10型耳聋相关的EYA4突变位点使用EYA4最长转录本(NM_004100.4)对以前报道的突变进行了重新分析。eyaVR代表可变区域,eyaHR代表同源区域,黑色表示插入/缺失/重复突变,绿色表示无义突变,蓝色表示剪切突变,橙色表示CNV,红色表示错义突变。Fig.2 Mutations of EYA4 associated with DFNA10 deafness The previously reported mutations were reanalyzed using the longest mRNA transcript of EYA4,NM_004100.4.EyaVR:variable region,eyaHR:homologous region,black:insertion/deletion/duplication mutation,green:nonsense mutation,blue:shear mutation,orange:CNV,red:missense mutation.
4 EYA4致聋的机制的探讨
基因突变可通过获能效应(gain of function)或失能效应(loss of function)导致ADNSHL,EYA4突变致聋是由于单倍体剂量不足导致的失能效应引起的。Zhang等利用小鼠Eya4HRR564X构建体进行了体外细胞实验,小鼠Eya4的突变R564X与人类的R587X是同源突变,Eya4HRR564X构建体转染到细胞内未能检测到相应截短蛋白的表达,且无法与Six1蛋白相结合,同样Eya4HR的其他不同类型截短构建体也在转染细胞内得到相同的结果,因此认为突变的EYA4蛋白因降解而单倍体剂量不足导致耳聋的发生[43]。EYA4致病还可能与内耳感觉细胞的Na+/K+-ATP酶功能异常有关,Wang等研究发现Na+/K+-ATP酶的亚单位atp1b2b在斑马鱼体内与eya4共表达具有时空一致性,eya4吗啡突变体斑马鱼内耳atp1b2b表达量下降,atp1b2b吗啡突变体与eya4突变体的表型相似,都出现毛细胞数量减少,半规管嵴发育不良及感觉运动异常,atp1b2b可挽回eya4突变体的听觉功能及结构异常。综上说明Na+/K+-ATP酶在斑马鱼发育过程中听觉功能上受eya4调节,eya4突变引起atp1b2b异常致耳聋发生,此研究在分子水平上为EYA4基因突变致耳聋发病的机制提供了一定可供参考的理论依据[6]。
利用小鼠模型探索人类遗传性耳聋的致病机制是一种常用的有效手段,但是Eya4敲除(Eya4-/-)的小鼠病变特点与DFNA10型耳聋不一致,Eya4-/-小鼠咽鼓管骨部变细、鼓口异位、纤毛数量减少,咽鼓管通气及排出功能异常导致分泌性中耳炎,同时伴有中耳畸形及严重的先天性听力损失,这与DF⁃NA10型耳聋迟发、渐进性的感音神经听力损失差别较大,在一些家系中儿童患者的分泌性中耳炎的发病或可借小鼠模型得以解释[44]。
5 EYA4致聋的临床遗传咨询
针对EYA4的致病突变进行遗传咨询可以帮助患者明确耳聋的病因,预估发病年龄、听力下降趋势及严重程度,预测耳聋的遗传规律及后代发病的风险。应注意由于迟发性发病的特点,新生儿听力筛查可能会通过,但要对EYA4突变者进行听力随访,及时发现听力下降并进行干预;对EYA4突变的患者应进行心脏相关病史询问、体检及检查,排除DCM的可能性;产前诊断和胚胎移植前遗传学诊断(preimplantation genetic diagnosis,PGD)是有效阻断EYA4垂直传递给下一代的方法,应在与患者充分沟通、合作的基础上,严格按照相关流程及规范完成[45]临床上在为EYA4患者提供合理指导建议的同时,还要兼顾疏导由基因检测结果产生的患者家庭的心理问题[46]。
6 小结和展望
EYA4突变引起的DFNA10型遗传性耳聋,临床表现可概括为迟发性的、影响中频听力的、进行性加重的感音神经性听力损失,但是表现度差异较大,目前报道的突变种类及家系数量较少,临床表型与基因型联系未发现明显的规律性。EYA4突变在韩国ADNSHL人群中所占比例较高,在中国人群报道的亦较多,但是中国人群发病比例尚待进一步统计研究,在中频受累的显性遗传性耳聋中,要考虑到EYA4致病的可能性。EYA4突变对心脏功能的影响存在较多疑惑,何种突变会影响心脏功能?突变是如何影响心脏发育和心脏功能的?是否存在其他基因修饰或起到更为主导的作用?更多临床表型的发现及基础的实验探索将会解答这些疑惑。EYA4致聋的机制与失能效应有关,可能通过调节内耳毛细胞Na+/K+-ATP酶影响听觉功能,构建合适的动物模型进行体内实验探究EYA4的致聋机制并探索可能的治疗方式如特定的药物治疗、基因治疗等是未来研究的重点,可利用CRISPR/Cas9技术精确编辑EYA4突变的哺乳动物模型进行相关研究。
