伴周围神经病变听神经病患者的临床特征分析
2019-03-04宋蒙涛王洪阳兰兰谢林怡熊芬王大勇关静王秋菊
宋蒙涛 王洪阳 兰兰 谢林怡 熊芬 王大勇 关静 王秋菊
解放军总医院耳鼻咽喉头颈外科解放军耳鼻咽喉研究所(北京100853)
听神经病(Auditory Neuropathy,AN),是一种由于听神经、内毛细胞和(或)突触受损引起听觉神经活动中断而导致的听觉障碍,患者表现为言语识别率明显低于纯音测听,听性脑干反应异常,外毛细胞功能正常[1]。听神经病分为非综合征型和综合征型,临床报道的听神经病患者以非综合征型多见,表现为单纯听力障碍不伴有其他神经系统病变,少数听神经病患者可伴发其他神经系统病变,如四肢麻木、肌张力低下、共济失调、视力下降等,具有多系统综合征表现,称为综合征型听神经病。Starr等[2]报道的10例听神经病患者中有8例随着病情发展出现其他周围神经症状。王锦玲等[3]在2011年报道了36例伴发神经系统疾病的听神经病患者。本文回顾性分析69例伴周围神经病变听神经病患者的临床特征,总结已报道的综合征型听神经病的遗传学与表型特征,并报告5个综合征型听神经病家系。
1 研究对象与方法
1.1 研究对象
本课题组2003年至2018年收集的531例听神经病病例,发现伴周围神经病变者69例。其中男性32例,女性37例;就诊年龄0.4-41岁,平均就诊年龄20.11±6.90岁;发病年龄0-26岁,平均发病年龄14.48±5.23岁;病程30天至20年不等,平均5.5年。家族性听神经病5例,婴幼儿(发病年龄≤3岁)听神经病4例,单侧听神经病1例;伴耳鸣者55例,伴前庭障碍者7例。本研究69例患者中均无噪声接触史、中耳疾病及明确的新生儿高危因素。
1.2 纳入标准与排除标准
纳入标准[4]:⑴ABR引不出或波形严重异常;⑵OAE或CM正常;⑶鼓室图正常,镫骨肌反射消失或阈值升高;⑷纯音测听以低频减退为主,主要累及125-1000Hz,多表现为轻、中度听力损失;⑸言语识别率不成比例地低于纯音听阈;⑹耳蜗电图表现为-SP/AP比值异常升高;⑺颞骨高分辨CT和/或磁共振成像(MRI)检查无异常发现;⑻存在视力下降、四肢末梢麻木、走路不稳等周围神经病变表现。排除噪声性聋、药物中毒性聋、突发性聋、梅尼埃病、听神经瘤等疾病。
1.3 研究方法
1.3.1 病史采集
由耳鼻咽喉科医师采用听神经病患者问诊专用问卷方式进行病史采集,包括:⑴基本信息:ID号、姓名、性别、年龄、问诊日期、分型诊断(非综合征听神经病/综合征型听神经病)、其他诊断(单侧听神经病、婴幼儿听神经病、家族性听神经病);⑵现病史:听力情况、听力下降治疗情况、耳鸣情况、眩晕情况、眼部症状、神经系统症状;⑶出生史:有无新生儿缺氧窒息、有无低出生体重、有无产期异常、分娩情况、有无新生儿高胆红素血症、有无其他新生儿疾病、有无母孕期耳毒性药物使用史;⑷既往史:有无噪声接触史、吸烟史、饮酒史、心脑血管病、高血脂、糖尿病、感染性或代谢性疾病、其他疾病、头颅外伤史及耳手术史;⑸家族史:有无耳聋家族史或其他疾病家族史、家系图;⑹血样采集情况。
1.3.2 听力学检查
应用Madsen Astera2型临床听力计,在标准隔音室内进行纯音测听和言语识别率检查,声信号的频率为125Hz-8kHz,以判断患者的听力损失程度及听力损失类型,以言语信号作为刺激声,通过让患者重复给出的言语信号来进行言语识别率检查;采用Madsen Capella测试仪,10DPOAE Probe插入式耳机行DPOAE检查,耳机频率响应范围设定在0.75kHz-8kHz,各频率信噪比≥6dB为DPOAE引出的标准;应用GSI TympStar型中耳分析仪进行声导抗检查,包括鼓室图测试和声反射测试;应用SmartEp测试仪进行听性脑干反应和耳蜗电图检查,最大给声强度为100dB nHL,以检测听觉神经传导功能;耳蜗电图记录三种电位,即动作电位(Ac⁃tion Potentials,AP)、耳蜗微音电位(Cochlear Micro⁃phonics,CM)、总和电位(Summting Potentials,SP)。
1.3.3 听力损失程度及听力图分型标准
根据世界卫生组织(WHO)1991年发布的听力损失分级标准[5],取纯音测听0.5、1、2、4kHz各频率的气导听阈平均值为平均听阈,进行听力损失程度分级,分级标准如下:正常(10-25dB HL),轻度(26-40dB HL),中度(41-60dB HL),重度(61-80dB HL),极重度(≥81 dB HL)。根据听力类型将纯音听力图分为5型,即峰型:极端频率(0.5或4kHz)听阈比中频(1-2k Hz)或高频差≥20 dB;平坦型:各倍频程听阈变化≤10 dB;上升型:每倍频程听阈减少≥5 dB;槽型:中频比极端频率听阈差≥20 dB;不确定型:不确定[6]。
1.3.4 统计学方法
应用SPSS 24.0统计处理软件进行统计学分析,连续变量表示为“平均值±标准差”,分类变量表示为“频数(百分比)”,并运用卡方检验进行比较。以P<0.05为差异有显著统计学意义。
2 结果
2.1 发病年龄及性别
本研究69例伴周围神经病变听神经病患者的初始发病年龄为0-26岁,平均发病年龄14.48±5.23岁,按照婴幼儿期(≤3岁)、儿童期(3-12岁)、青少年期(12-18岁)和成人(≥18岁)四个年龄段对该组患者的男女发病年龄进行统计,其中男32例(46.38%),女37例(53.62%),男女比例为1:1.16,性别构成差异无统计学意义(P>0.05);婴幼儿期发病4例,占5.79%;儿童期发病10例,占14.49%;青少年期发病36例,占52.17%;成人发病19例,占27.54%。
2.2 临床症状特点
本研究69例患者的周围神经病变表现主要包括视力下降和四肢末梢麻木、走路不稳、四肢运动障碍等感觉运动障碍,其中伴视力下降者48例,占69.57%;伴感觉运动障碍者32例,占46.38%;同时伴有视力下降和感觉运动障碍者7例,占10.14%。以听力下降、言语识别率降低为首发症状者有44例(63.77%),其余25例(36.23%)为首先出现视力下降、四肢麻木、走路不稳等周围神经病变表现。54例(78.26%)患者伴有耳鸣,7例(10.14%)患者伴有眩晕或前庭功能障碍。(表1)

表1 不同发病时期患者的临床症状特点Table 1 Characteristics of clinical symptoms in patients with different onset period

图1 伴周围神经病变听神经病患者不同类型纯音听力图(%)Fig.1 Different types of audiogram in patients with auditory neuropathy accompanying peripheral neuropathy(%)
2.3 听力学表现
2.3.1 纯音测听
本研究69例伴周围神经病变患者中65例(130耳)进行了纯音测听检查(其余4例为婴幼儿听神经病患者,纯音测听无法配合):纯音听力图表现为上升型87耳,占66.92%;峰型14耳,占10.77%,波峰出现在1或2kHz处;平坦型13耳,占10%;不确定型16耳,占12.31%(图2)。听力损失程度分级:平均听阈正常28耳,占21.54%,但均伴有低频听力下降;轻度听力损失43耳,占33.08%;中度听力损失46耳,占35.38%;重度听力损失10耳,占7.69%;极重度听力损失3耳,占2.31%。(表2)

图2 综合征型听神经病患者家系图(5例)Fig.2 Family trees of patients with syndrome auditory neuropathy(5 cases)
2.3.2 听性脑干反应(ABR)
本研究69例(138耳)伴周围神经病变的听神经病患者中,仅1例右耳可见Ⅰ波,潜伏期1.55ms;1例Ⅰ-III波重复性差,V波潜伏期6.25ms;其余均自Ⅰ波起未引出(>100dB SPL)。
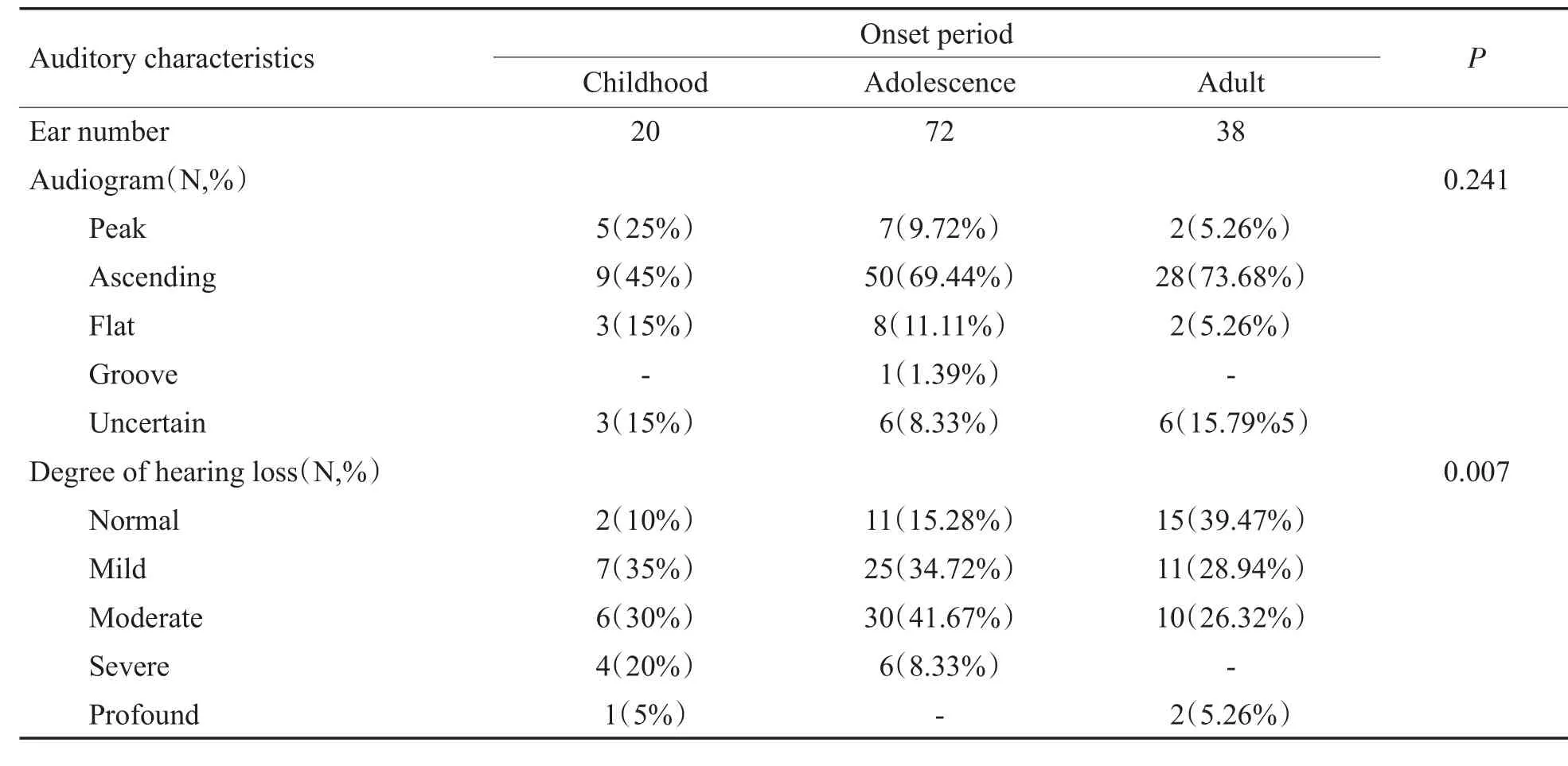
表2 不同发病时期患者的纯音测听结果Table 2 The results of pure tone test in patients with different onset period
2.3.3 声导抗测试
本研究68例(136耳)患者行声导抗测试,鼓室图7耳呈“As”型,2耳呈“C”型,5耳呈“Ad”型,其余均为“A”型曲线;镫骨肌声反射54例(79.41%)同侧及交叉声反射均未引出,10例仅1或2个频率可引出反应,2例单侧引出对侧引不出,1例两侧均能引出。
2.3.4 畸变产物耳声发射(DPOAE)
69例(138耳)患者均行DPOAE检查,其中2耳DPOAE多数频率引不出而CM波存在,31耳个别频率引不出,其余105耳各频率均可引出。
2.3.5 耳蜗电图(ECochG)
69例患者中25例(47耳)进行了耳蜗电图检查,对双耳―SP和AP的潜伏期、幅值以及―SP/AP的值进行记录,结果如表3所示。
2.3.6 言语识别率
69例患者中44(85耳)例行言语识别率测试(SDS),最大言语识别率≥90%有9耳,占10.59%;71%-89%有13耳,占15.29%;51%-70%有14耳,占16.47%;≤50%有49耳,占57.65%。言语识别率下降程度明显差于纯音听阈。
3 综合征型听神经病家系报告
3.1 0702703家系
吉林省家系,姐姐和弟弟患病,听力学表现符合听神经病,同时伴耳鸣、视力下降、肌无力等症状。弟弟为先证者(Ⅱ5),24岁,双耳中度至重度听力损失,13岁时开始听力下降,进而出现耳鸣、视力下降、双下肢无力、走路不稳及严重肌萎缩,呼吸肌受累后死亡。其姐姐(Ⅱ3)31岁,11岁时出现听力下降、耳鸣和视力下降,但症状较轻。
3.2 0501405家系
河北省家系,哥哥和妹妹患病,2人均表现为典型的听神经病症状和视力下降。妹妹为先证者(Ⅱ3),18岁,16岁时出现听力下降,家长诉其听力下降后能听到声音,但不理解语义。哥哥(Ⅱ1)表型与妹妹相似,16岁左右出现听力下降,嘈杂环境下言语识别率明显下降。
3.3 0804895家系
山东省家系,哥哥和妹妹患病,2人均诊断为伴听神经病的Charcot-Marie-Tooth(CMT)综合征/腓骨肌萎缩症,听力学表现符合听神经病,且伴有耳鸣、四肢感觉运动障碍。哥哥为先证者(Ⅱ1),24岁,22岁时出现言语识别率下降,轻度听力损失伴耳鸣,下肢麻木感且走路不稳。妹妹(Ⅱ2)18岁,表型与哥哥相似,12岁时发病,左耳重度听力损失,右耳中重度听力损失,且走路不稳症状较哥哥重。
3.4 0602403家系
山西省家系,家系中有4人患病,包括3名男性患者及1名女性患者,发病年龄均在10-20岁之间,诊断为伴听神经病的Charcot-Marie-Tooth(CMT)综合征/腓骨肌萎缩症,听力学表现符合听神经病,伴有耳鸣、走路不稳、四肢感觉障碍、肌张力低下、共济失调等表现。先证者(Ⅲ9)为男性,36岁,双耳重度听力损失,14岁时开始出现听力下降,言语识别率下降,伴有四肢末梢神经感觉异常,双下肢肌张力低下,深感觉减退。其哥哥(Ⅲ1)16岁时出现明显听力障碍同时伴有耳鸣,言语识别能力差,走路不稳等表现。弟弟(Ⅲ11)11岁时发现言语识别力差,16岁时出现轻度听力下降,也具有平衡能力差及走路不稳等表现。
3.5 1007170家系
辽宁省家系,家族中7人患病,包括6名男性及1名女性,发病年龄为7-18岁,均表现为典型的听神经病特征,并伴有不同程度的耳鸣、四肢麻木或肌无力,临床诊断为听神经病伴周围神经病变。先证者(Ⅳ1)为男性,20岁,1年前出现双耳听力下降伴耳鸣、四肢麻木感,磁共振检查显示双侧听神经纤细。
4 讨论
听神经病是一种特殊的听觉障碍疾病,可以独立发病,也可以是其他周围神经病或遗传性疾病所伴发的听神经局部病变,本课题组收集的531例听神经病病例中,先后伴发周围神经病变者有69例,占12.99%。听神经病好发于3岁以前的婴幼儿和12-20岁的青少年,30岁以上者少见[7];本研究69例伴周围神经病变的听神经病患者中有55例发病年龄在12-26岁,占79.71%,婴幼儿患者4例,无30岁以上发病者。综合征型听神经病的听力学表现可以是首发症状,也可以同时或晚于其他周围神经病变表现,本研究69例伴周围神经病变的听神经病患者中,以听神经病为首发症状者有44例(63.77%);王锦玲等[3]在2011年报道的36例伴发神经系统的听神经病患者中,有22例(61.11%)患者首先出现听力下降,言语识别率降低。齐悦等[8]在2013年的研究结果显示,综合征型听神经病患者的耳鸣发生率明显高于非综合征型听神经病患者(84.00%>42.00%),本研究69例伴周围神经病变的听神经病患者中,伴发耳鸣者54例,占78.26%。

表3 听神经病患者(25例,47耳)耳蜗电图结果(x±s)Table 3 Results of electrocochleogram in patients with auditory neuropathy
周围神经病是一组常见的由周围神经受累导致的神经疾病,主要表现为四肢麻木、腱反射减弱、肌张力低下、共济失调、视力下降等[9]。本组听神经病患者伴发的周围神经病变主要表现为视力下降和四肢末梢麻木、走路不稳、四肢运动障碍等感觉,其中伴视力下降者48例(69.57%),伴感觉运动障碍者32例(46.38%),同时伴有视力下降和感觉运动障碍者7例(10.14%),可见视力下降在伴周围神经病变的听神经病患者中具有较高的发生率。研究发现,由OPA1基因中Arg445His错义突变导致的常染色体显性视神经萎缩相关听力障碍通常伴有进展缓慢的视力下降,且听力学评估表现为听神经病[10];此外,Leber遗传性视神经病[11]、Wolfram 综合征[12]、Mohr-Tranebjaerg综合征[13]、Ref⁃sum’s病[14]等综合征型听神经病均可伴发不同程度的视力下降。
听神经病是一种病因复杂多样的异质性疾病,其病因可以是先天性的,也可以是后天获得性的。获得性听神经病的病因包括早产、高胆红素血症、缺氧、低氧、先天性脑畸形、围产期颅内出血、窒息、耳毒性药物、脱髓鞘疾病(如多发性硬化症)以及AIDS或HIV感染[15];相反,先天性听神经病主要是由于基因异常引起的,可以单独发病,也可以伴有其他综合征(如上文所述)。据研究报道,大约40%的听神经病与遗传因素有关[16]。Starr等[17]按照遗传方式将听神经病分为常染色体显性遗传、常染色体隐性遗传、X连锁隐性遗传以及线粒体DNA突变等。目前报道的综合征型听神经病多为散发病例,本文报告了5例家族性听神经病患者,绘制了家系系谱图(图2),在此基础上根据家系成员的表型特征进行遗传学分析。结果显示,0804895家系与0702703家系具有常染色体隐性遗传特征:⑴发生与性别无关,男女发病机会均等;⑵多为散发,通常无连续传递现象;⑶患者双亲表型往往正常,但均为致病基因携带者;⑷近亲结婚发病率明显增高。0602423家系与1007170家系具有X连锁隐性遗传特征:⑴男性患者远多于女性患者;⑵双亲无病时,儿子可能发病,女儿则不会发病;儿子如果发病,母亲肯定为携带者;⑶女性患病,其父一定为患者,母亲一定是携带者。
近年来,伴周围神经病变的综合征型听神经病的诊断率不断提高,目前已发现多种综合征型听神经病,包括腓骨肌萎缩症(CMT)、Leber遗传性视神经病(LHON)、常染色体显性遗传性视神经萎缩(ADOA)、常染色体隐性遗传性视神经萎缩(AROA)、Friedreich’s共济失调、Mohr-Tranebjaerg综合征(DDON/MTS)、Wolfram综合征、Refsum病、染色体病等,这些综合征多为单基因遗传病,遗传因素在疾病的发病中起到了重要作用[18],目前已发现 PMP22、MPZ、NF-L、NDRG1、GJB1、GJB3、OPA1、TMEM126A、FXN、TIMM8A、WFS1、AIFM1、CACNA1D、PAHX基因以及线粒体突变12SrRNA(T1095C)和MTND4(11778mtDNA)等10余种与综合征型听神经病相关致病基因[4],本文对目前已报道的综合征型听神经病的遗传学及表型特征进行了总结,见表4。
后续随访及研究计划:本研究纳入的69例患者中,仍有部分患者检查结果不够完善,其周围神经病病变的临床诊断主要依据临床症状和神经内科相关辅助检查做出,未明确评价标准及程度判定缺乏病理活检及基因诊断等针对性检查,因此在今后的随访工作中,需继续完善患者的随访复诊流程,安排专门人员对患者进行电话随访,并向其普及综合征型听神经病的发病特征,就其周围神经病变表现向患者提出合理的专科会诊建议,进一步明确病因,揭示综合征型听神经病的发病机制,进而采取个性化治疗,使患者得到最大的收益。
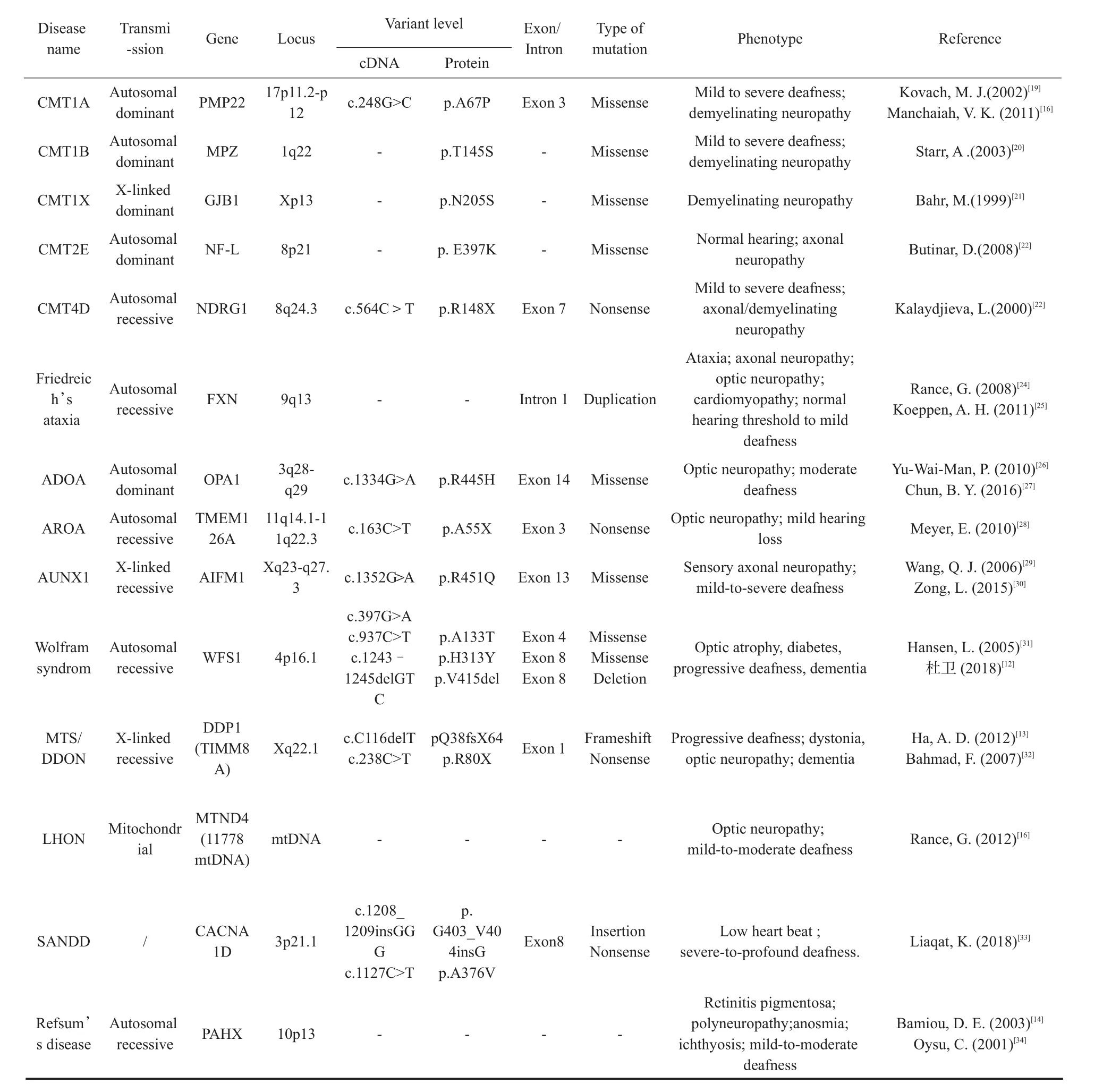
表4 目前已知综合征型听神经病的遗传学和表型特征Table 4 The genetic and phenotypic characteristics of the syndrome auditory neuropathy
