非综合征型听神经病患者OTOF基因筛查研究
2019-03-04梁鹏飞王锦玲王剑王淑娟邱建华查定军
梁鹏飞 王锦玲 王剑 王淑娟 邱建华 查定军
空军军医大学西京医院耳鼻咽喉头颈外科
听神经病(auditory neuropathy,AN)是一种有特 殊临床表现的听力障碍性疾病。最早由Starr等[1]提出,认为自耳蜗至进入脑干之前的第VIII脑神经听支(耳蜗神经)受损。主要的临床表现包括:反应内毛细胞及听觉传导通路功能的听性脑干反应(auditory brainstem response,ABR)缺失或严重异常,而主要反应外毛细胞功能的诱发性耳声发射(evoked otoacoustic emission,EOAE)正常或基本正常,言语识别率显著差于纯音听阈水平等。王锦玲等[2]对286例听神经病患者进行临床流行病学调查统计,结果显示听神经病患者约占同期听力减退患者的1.37%,而在各种原因所致的ABR异常的聋病患儿中发病率高达11%[3]。
目前听神经病的发病机理不明,随着相关基因的不断发现,开始从分子水平上揭示听神经病谱系障碍的病理机制。OTOF基因是非综合征型隐性遗传性感音神经性耳聋(DFNB9)的致病基因,也是最早确定的与非综合征型隐性遗传性听神经病相关的基因[4]。近年来,研究学者们在听神经病家系及散发患者中,广泛开展了OTOF基因的筛查研究,报道了多个可引起听神经病表型的病理性突变位点,但国内尚缺乏散发听神经病患者的大样本OTOF基因突变检测的临床研究。本课题收集248例听神经患者进行OTOF基因全部编码区序列检测,以期明确与我国听神经病相关OTOF基因的高发突变位点。
1 资料与方法
1.1 研究对象
本研究中的听神经病患者的临床资料和外周血样本均来自于空军军医大学第一附属医院耳鼻咽喉头颈外科聋病基因诊断中心。所收集到到的病例主要来自中国西部及中部地区。本研究中的受检者包括2009至2018年收集的听神经病患者248例,其中男性126例,女性122例,年龄1岁至44岁,平均年龄22.51±9.94岁。全部患者进行纯音听阈测试、声导抗、耳声发射(DPOAE)、听性脑干反应(ABR)、言语识别率、眼震电图(ENG)、前庭诱发肌源电位(VEMP)以及颞骨CT扫描,婴幼儿行听性稳态反应检查。每位患者均进行详细的病史采集,包括基本信息、耳聋病史、个人史、母亲孕期身体状况、用药史、外伤史、家族史等。排除与国外种族联姻家族或近亲联姻家族,以及有外耳、中耳畸形患者。有家族史者只计入先证者。前期工作筛查常见耳聋基因(GJB2基因、SLC26A4基因、线粒体12SrRNA1555以及1494位点)均为阴性。同时选取100名地域相当的听力正常人作为对照组。
1.2 听神经病患者临床症状诊断标准[5]
1、纯音听阈以低频减退为主;2、言语识别率不成比例地差于纯音听阈;3、镫骨肌声反射同侧及交叉声反射不能引出或阈值升高;4、ABR波形缺失或严重异常;5、DPAOE和(或)微音电位(CM)正常;6、影像学检查(颞骨CT和/或内听道MRI)正常。
1.3 血液DNA提取
患者或其监护人签署知情同意书后,抽取被检测者外周静脉血3-5ml,EDTA抗凝,采用康为世纪中量全血基因组提取试剂盒提取基因组DNA,使用分光光度计检测纯度,满足OD260/280=1.7-2.0,取适量样本稀释至浓度100-200ng/µl,剩余置-80℃保存。
1.4 引物设计
参照美国国立生物技术信息中心(NCBI)基因数据库中人类OTOF基因标准序列(基因ID:9381)设计引物,扩增全部编码区及剪切位置,见表1。
所有引物均使用Primer3在线软件设计,由西安擎科泽西生物技术有限公司合成。
1.5 PCR反应体系
PCR反应采用50µl体系,包括2Pfu PCR Mas⁃terMix 25µl,上下游引物各 1µl,DNA 模板 2µl,ddH2O 21µl,PCR扩增条件采用Touchdown方法,反应条件:95℃预变性4min,95℃变性30s,62℃起始退火30s,72℃延伸30s,12个循环,每个循环降低0.5℃;随后95℃变性30s,56℃退火30s,72℃延伸30s,30个循环;72℃再延伸10min后,4℃保存。如图1所示。

图1 PCR反应条件Fig.1 PCR condition
1.6 基因序列比对及相关生物学分析
248例被检测对象的基因扩增产物经纯化后,使用ABI 3700 DNA测序仪进行测序,测序引物与PCR扩增引物相同。结果使用Chromos软件读取并利用Seqman软件与NCBI数据库提供的标准序列进行比对分析。
2 结果
本研究中共设计35对引物,涵盖了OTOF基因的所有48个外显子及其侧翼区域,扩增片段与预期一致。将测序结果与NCBI数据库中的标准序列(NM_194248.2)进行比对,共在20例听神经病患者中发现9种可能致病的突变方式,包括7种错义突变和2种剪切方式的改变(表2),OTOF基因突变检出率为8.06%(20/248)。同时还检出4种已有文献报道的多态性改变(表3)。9种可能的致病突变方式中,位于40号外显子的c.5026C>T检出率最高(6/248),共有7人存在Exon40的突变。结果显示,248名听神经病患者OTOF突变集中在Exon40、In⁃tron32、Exon12、Exon18、Exon24、Intron28和 Exon7中(依检出数目由高至低)。

表1 OTOF基因扩增引物Table 1 Sequence of primers used for OTOF amplification
本研究在1例儿童听神经病患者的基因组中,检测到OTOF基因发生c.2977-2978delAG/c.3570+2T>C复合杂合突变。该名患者首次就诊年龄7岁,男童,听力测试结果(图2)显示:双侧极重度感音神经性听力损失;声导抗检测双耳均为As型曲线,双耳声反射阈均未引出;右耳2-8kHz可引出DPOAE,左耳0.75-8kHz可引出DPOAE;ABR阈值双耳>100dBnHL;双侧颞骨CT及内听道MRI均未见异常。

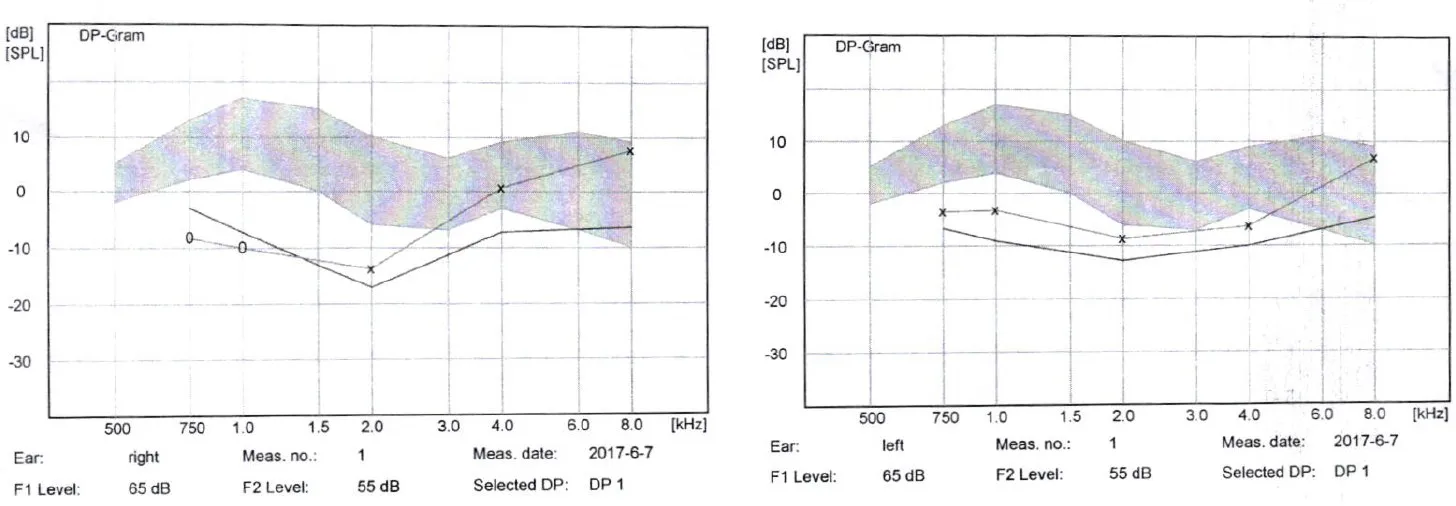
图2 携带OTOF c.2977-2978delAG/c.3570+2T>C突变患者听力学表现Fig.2 Audiological performances of the patient with OTOF c.2977-2978delAG/c.3570+2T>C mutation

表2 248名患者OTOF基因可能致病突变统计Table 2 Probable-pathogenic variants of OTOF identified in 248AN patients

表3 OTOF基因多态性改变Table 3 polymorphic changes of OTOF
3 讨论
人类OTOF基因位于2p23.3,基因DNA序列全长101496bp,共有4种长短不同的转录变异体,其中最长的亚型包含48个外显子,其中外显子1未被编码,编码长度为1997个氨基酸的OTOFER⁃LIN蛋白。
OTOF基因是1999年由Yasunage等[12]定位并命名,并被认为是常染色体隐性遗传性非综合征型耳聋(DFNB9)的致病基因。2003年Varga R等[4]通过连锁分析及突变检测的方法,证明OTOF基因突变是导致非综合征型隐性听神经病家系的致病原因。免疫荧光染色结果显示,胚胎期小鼠的耳蜗内毛细胞、外毛细胞以及螺旋神经节部位均可检测到OTOFERLIN蛋白的表达,而在成熟小鼠的耳蜗,OTOFERLIN蛋白集中于内毛细胞基底外侧部以及带状突触小泡和突触前膜上。OTOF基因敲除小鼠呈现出功能性外毛细胞以及听神经,推测重度听力障碍是由于内毛细胞的功能缺陷所致[13]。
近年来,在非综合征型听神经病患者中开展OTOF基因筛查已成为疾病分子病因学研究的热点课题,至今在不同地区和人群中报道了多个可引起听神经病表型的病理性突变位点[6-11]。本课题在248例的听神经病患者中,筛查OTOF全部编码区序列,共检出9种OTOF可能致病方式。这些突变集中在Exon40、Intron32、Exon12、Exon18、Exon24、In⁃tron28和Exon7中。在门诊耳聋患者或者听力正常人群中,开展相关外显子的筛查,将对携带者的婚育具有指导意义。我们的研究发现携带这些突变的患者多为单杂合形式存在,仅有1例患者为OTOF基因复合杂合突变携带者。既往的文献表明,OTOF基因是一种隐性致病基因,单等位基因突变尚无法确定其致病性,只有发生双等位基因突变才会出现听神经病症状,说明OTOF基因并不是大多数受检患者的唯一患病原因。随着对听神经病的深入研究和临床听力学检测技术的进步,目前听神经病的发病率高于以往的报道。而基因检测手段的飞速发展,越来越多的基因被认为与听神经病相关。迄今为止,发现的非综合征型听神经病相关的基因除了OTOF基因以外,还包括PJVK、DIAPH3、SLC17A8、DIAPH3等[14],王秋菊教授团队在国际上首次报道了一个X-连锁隐性遗传性听神经病基因作为(AUNX1)基因座[15],并发现AIFM1基因具有高度相关性[16]。在接下来的工作中,我们将继续进行上述基因的检测研究,以期丰富听神经病相关基因的突变筛查谱。
