PI3K/Akt/GSK-3β通路在体外培养神经元缺氧缺血损伤中的作用
2019-02-16赵婧,李强,何玲
赵 婧,李 强,何 玲
(川北医学院附属医院新生儿科,四川南充 637000)
新生儿缺氧缺血性脑病(HIE)是围产期窒息严重并发症之一,病死率及致残率高[1-2]。目前认为大脑缺氧缺血(hypoxia-ischemia,HI),影响神经突极性结构,破坏神经网络的形成,可能是引起HIE神经系统后遗症的重要原因之一[3]。迄今,已发现神经元极性分子糖原合成激酶-3β(glycogen synthase kinase-3β,GSK-3β)及磷脂酰肌醇-3激酶/丝氨酸/苏氨酸蛋白激酶(phosphatidylinositol-3 kinase/Akt,PI3K/Akt)在神经突极性建立与维持中发挥重要作用[4-5]。在HI神经元中,PI3K/AKT/GSK-3β信号通路是否参与神经元损伤的调节及其信号传递的机制等尚不清楚。因此,本研究在建立新生大鼠皮质神经元氧糖剥夺(oxygen and glucose deprivation,OGD)模型,模拟HI基础上,应用PI3K/Akt抑制剂LY294002和GSK-3β抑制剂SB415286进行干预,检测Akt、p-Akt(Ser-473)、GSK-3β、p-GSK-3β(ser-9)的表达变化及神经突形态、长度变化,明确PI3K/Akt/GSK-3β信号通路在HI神经元损伤中的作用,现报道如下。
1 材料与方法
1.1试剂 LY294002、SB415286购于美国Cell signaling公司,兔抗鼠Akt、p-Akt (ser-473)抗体、兔抗鼠GSK-3β、p-GSK-3β(ser-9)抗体、小鼠微管蛋白TUJ1单克隆抗体购于美国Millipore公司,小鼠β-actin单克隆抗体购于美国Santa Cruz公司,辣根过氧化物酶(HRP)标记羊抗鼠IgG、HRP标记羊抗兔IgG购于北京中杉金桥生物技术有限公司,BCA蛋白定量试剂盒购于美国Chemicon公司,磷酸酶抑制剂、化学发光试剂盒购于美国Chemicon公司,Western blot一抗、二抗去除液购于碧云天公司。
1.2方法
1.2.1原代神经元培养 新生SD大鼠在75%乙醇中浸泡3~5 min,无菌条件下断头杀死,取出大脑。在解剖镜下分离出大脑皮质并剪碎,以0.25% 胰蛋白酶和终浓度为10 μg/mL 的DNAseⅠ消化,通过200目细胞筛网过滤后,1 000 r/min离心5 min,弃上清液,加含2% B27细胞培养添加剂、1%谷氨酰胺的无血清神经培养基吹打成单细胞悬液,以2×106/mL的细胞密度接种于多聚L赖氨酸(PLL)包被的6孔板盖玻片或25 cm2培养瓶中,置37 ℃,5% CO2孵箱中培养2 d后半量换液,培养至第7天用于实验。
1.2.2体外模拟HI 建立体外OGD模型[6]。将原代培养至第7天的神经元弃去培养液,加入无糖DMEM培养液模拟细胞缺血状态,再将细胞放入3气培养箱中,用含95% N2、5% CO2混合气从进气管充气,培养3 h。3 h后终止缺氧,并更换为含葡萄糖的神经元培养基,以形成再灌注。取再灌注后0.5、3、6、12、24、48 h不同时间点的神经元进行实验。
1.2.3实验分组 实验分为4组:(1)正常对照组:正常培养液孵育的神经元;(2)OGD组:模型制备后,取再灌注0.5、3、6、12、24、48 h观察;(3)LY294002干预组:应用LY294002(20 μmol/L)预处理神经元12 h,再以相同浓度在OGD中维持3 h后更换为含葡萄糖的神经元培养基;(4)SB415286干预组:应用SB415286(40 μmol/L)预处理神经元12 h,再以相同浓度在OGD中维持3 h后更换为含葡萄糖的神经元培养基。
1.2.4Western blot检测各组神经元Akt、p-Akt、GSK-3β、p-GSK-3β蛋白表达 冰上提取神经元蛋白后,BCA法测定蛋白浓度,-80 ℃保存。等量的样品蛋白,经十二烷基硫酸钠聚丙烯酰胺凝胶电泳(SDS-PAGE)分离后,转膜,相继加入相应的一抗及二抗进行免疫反应,ECL发光剂显色,置于Omega 12 ic凝胶成像仪中显像并拍照,应用NIH图像测其吸光度(OD)值,以目的蛋白条带OD值/β-actin条带OD值来表示目的蛋白表达水平。
1.2.5神经突免疫荧光染色 采用免疫荧光标记TUJ1。6孔板培养神经元,4%多聚甲醛4 ℃固定20 min,磷酸缓冲盐溶液(PBS)洗3次后滴加免疫荧光封闭液,4 ℃孵育30 min,滴加TUJ1一抗(1∶200),4 ℃孵育过夜;PBS洗3次后滴加异硫氰酸荧光素(FITC)标记二抗(1∶200),37 ℃孵育40 min;滴加4′,6-二脒基-2-苯基吲哚(DAPI),常温孵育5 min;封片后荧光显微镜下镜检。

2 结 果
2.1OGD后Akt、p-Akt、GSK-3β、p-GSK-3β蛋白表达 总Akt(F=1.214,P>0.05)及总GSK-3β(F=0.376,P>0.05)的表达在OGD后未发生变化。p-Akt在OGD后0.5 h明显降低,在经过短暂升高后(OGD后3、6 h),在12 h开始下降,24 h降至最低(F=525.33,P<0.05)。p-Akt/Akt值,在OGD后24 h降至最低(F=674.46,P<0.05),见图1。p-GSK-3β在OGD后0.5 h开始降低,24 h降至最低(F=557.243,P<0.05)。p-GSK-3β/GSK-3β值在OGD后24 h降至最低(F=376.581,P<0.05),见图1。
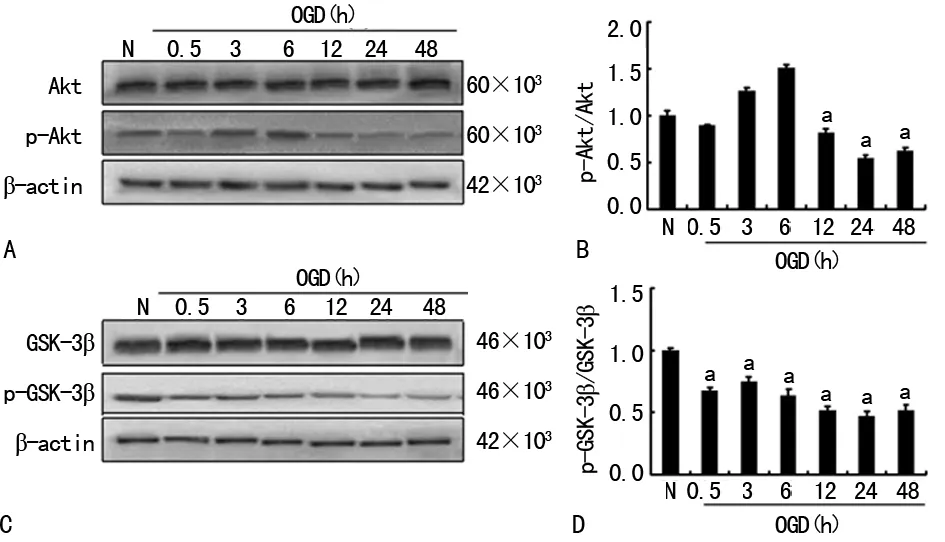
A:皮质神经元OGD后Akt、p-Akt表达的Western blot图;B:定量分析p-Akt/Akt;C:皮质神经元OGD后GSK-3β、p-GSK-3β表达的Western blot图;D:定量分析p-GSK-3β/GSK-3β;N:正常对照组;a:P<0.05,与对照组比较

A:Akt和p-Akt蛋白表达的Western blot图;B:定量分析p-Akt/Akt;C:GSK-3β和p-GSK-3β蛋白表达的Western blot图;D:定量分析p-GSK-3β/GSK-3β;1:OGD 6 h组;2:正常对照组;3:LY294002干预组;a:P<0.01,与OGD组比较
2.2LY294002抑制Akt、GSK-3β磷酸化 Western blot结果显示,LY294002干预后,总Akt和GSK-3β并未发生变化,而p-Akt和p-GSK-3β表达减少(P<0.05)。与OGD 6 h组比较,p-Akt/Akt及p-GSK-3β/GSK-3β在应用LY294002后分别降低了约49%(F=242.263,P<0.01)和42%(F=128.114,P<0.01),而OGD 6 h组和正常对照组之间比较差异无统计学意义(P>0.05),见图2。
2.3SB415286促进GSK-3β磷酸化 Western blot结果显示,SB415286干预后,总Akt、p-Akt和总GSK-3β并未发生变化,而p-GSK-3β表达增加(P<0.05)。与OGD 6 h组比较, p-GSK-3β/GSK-3β在应用SB415286后增加了37% (F=122.347,P<0.01),而OGD 6 h组和正常对照组之间比较差异无统计学意义(P>0.05),见图3。
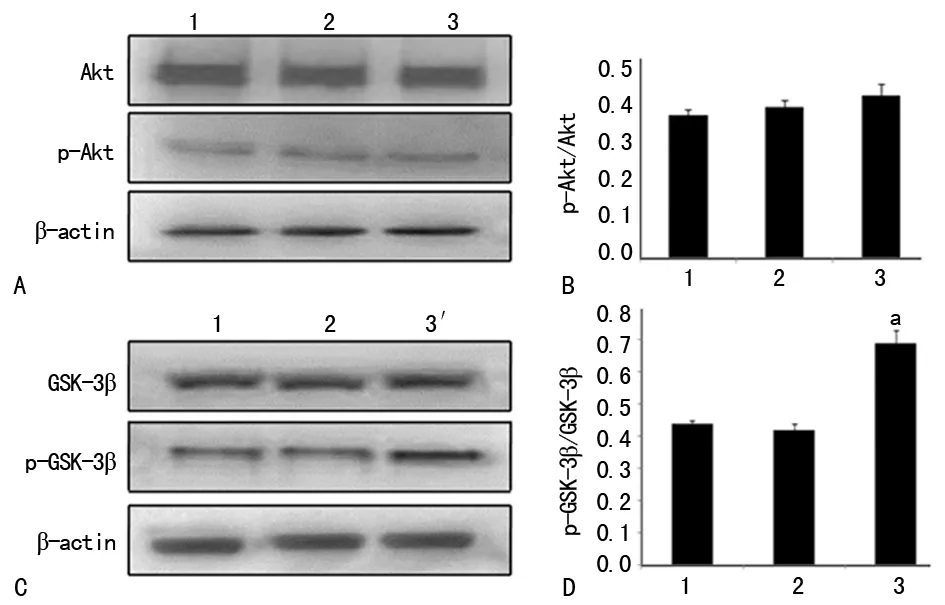
A:Akt和p-Akt蛋白表达的Western blot图;B:定量分析p-Akt/Akt;C:GSK-3β和p-GSK-3β蛋白表达的Western blot图;D:定量分析p-GSK-3β/GSK-3β;1:OGD 6 h组;2:正常对照组;3:SB415286干预组;a:P<0.01,与OGD组比较
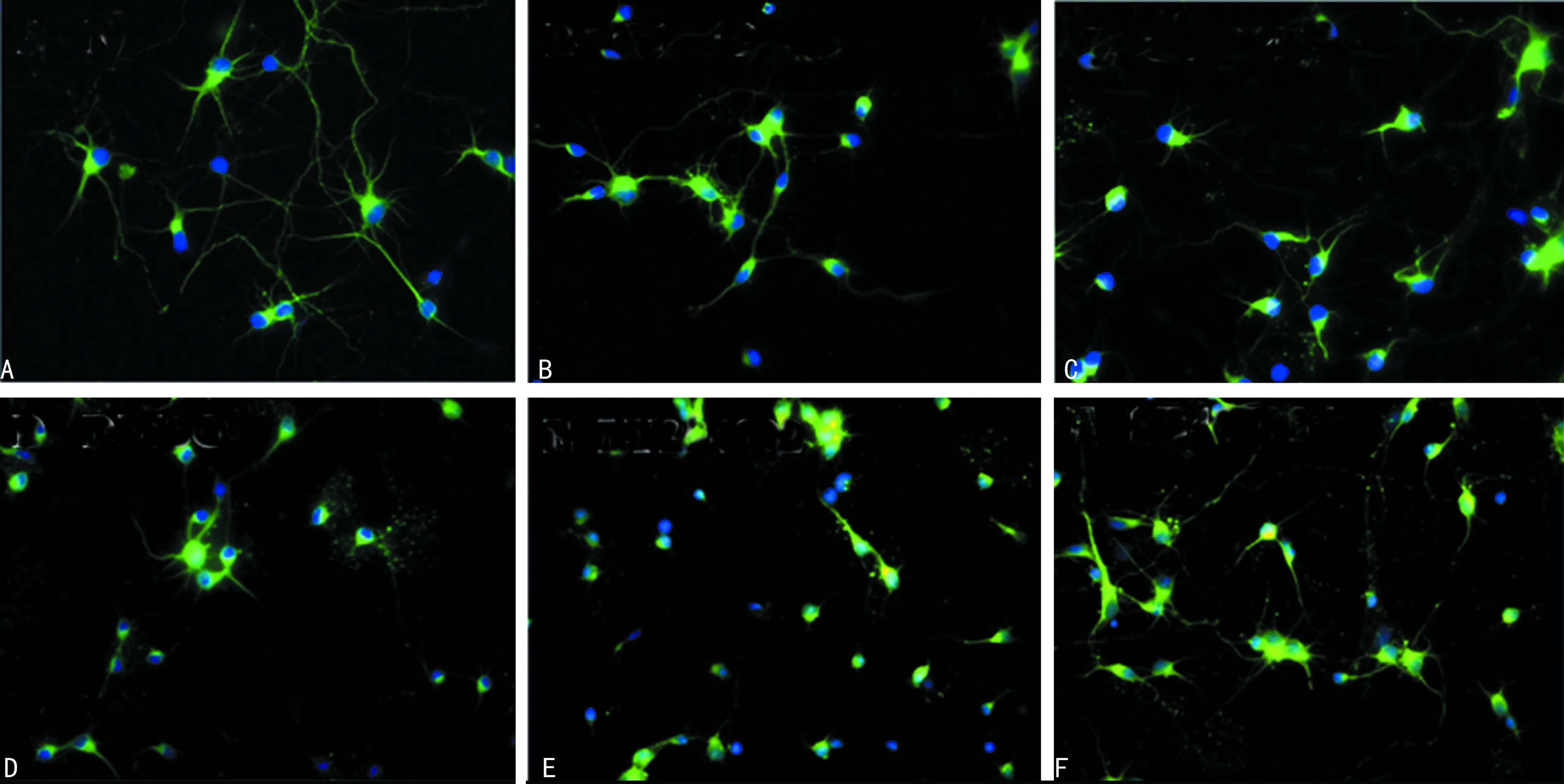
A:培养第7天神经元;B:OGD后24 h组神经元;C:OGD后48 h组神经元;D:正常对照组神经元;E:LY294002干预组神经元(OGD后48 h);F:SB415286处理组神经元(OGD后48 h)
2.4抑制PI3K/Akt活性加重HI后神经元神经突损伤 如图4所示,绿色荧光标记的是TUJ1,蓝色标记的是细胞核。培养至第7天的神经元形成成熟的极性结构,即数个树突及1个轴突染成绿色荧光,采用Neurocida软件测量轴突平均长度为(5 287±262) μm。OGD后24 h,神经元胞体及神经突近端肿胀,可见固缩或碎裂的细胞核轴突平均长度为(2 985±225)μm。48 h后神经突变细、变直、断裂,分支减少,轴突长度为(1 086±67)μm。以OGD后形态变化较明显的时间点48 h作为对照,LY294002干预组在OGD后48 h,神经元极性结构消失,仅可见围绕胞体短细的突起,平均长度为(445±85)μm。应用LY294002后明显加重神经突损伤,轴突长度较OGD 48 h组差异有统计学意义(P<0.01)。
2.5抑制GSK-3β活性减轻缺氧缺血后神经元神经突损伤 SB415286处理组在OGD 48 h后,神经突近端稍有肿胀,远端稍变细,分枝稍减少,与OGD 48 h组[(1 086±67)μm]比较轴突长度明显更长,平均长度为(2 692±207)μm,见图4F。应用SB415286后可减轻神经突损伤,轴突长度较OGD 48 h组差异有统计学意义(P<0.01)。
3 讨 论
神经突是神经元传递和接受神经信号冲动的基本结构,也是神经元极性结构形成的基础。在许多神经退行性疾病中都涉及神经突结构和神经元极性的损伤[7-9]。因此,探索引起神经突损伤的分子机制及实施相应的干预措施十分重要。本研究发现,在体外培养神经元OGD后24 h形态已经发生明显变化,神经突变短,OGD后48 h神经突分支减少、断裂。这种结构改变可能与HI后不可逆的神经元功能损伤有关。
在对神经元极性形成和维持的分子生物学研究中发现,高活性的Akt(Myr-Akt)能促进轴突生长[4]。并且通过上调PI3K增强Akt磷酸化后可促进外周神经再生[5]。PI3K/Akt通过磷酸化调节相关蛋白等途径促进细胞生长、抑制细胞凋亡[10]。本研究发现,p-Akt在OGD后0.5及12 h明显降低,并在后续时间点持续低表达,提示与相应时间点神经元结构损伤相关。本研究同时发现在OGD后3、6 h p-Akt有短暂性升高,这可能与HI早期的神经元试图通过上调Akt活性减轻损伤有关[11],但这种短暂性的Akt活性改变并不能逆转神经突结构损伤。
GSK-3β是一种多功能的丝氨酸/苏氨酸类蛋白激酶[12]。GSK-3β在神经突的磷酸化状态对神经元极性结构形成起决定作用[13]。神经元共转染高活性的GSK-3β(GSK-3βS9A)和高活性的Akt(Myr-Akt)后,可逆转Myr-Akt诱导形成的多根轴突[14]。本研究发现神经元OGD后p-GSK-3β的表达持续降低,这种变化可能与神经突损伤相关。同时发现在OGD后12 h p-Akt/Akt、p-GSK-3β/GSK-3β的比值变化趋势基本一致,表明两者可能存在一定联系。因此进一步检测抑制PI3K/Akt或GSK-3β活性,探讨3个蛋白间的调控关系。
LY294002和SB415286分别是PI3K/Akt和GSK-3β的特异性抑制剂[15-16]。抑制PI3K/Akt活性后,神经元p-GSK-3β表达降低。而抑制GSK-3β后,神经元Akt及p-Akt的表达均未发生变化,表明参与OGD后神经元损伤的分子调节中,GSK-3β是PI3K/Akt的下游信号分子。同时LY294002组神经突在OGD后48 h极性结构消失,表明抑制PI3K/Akt活性明显加重神经突损伤。而SB415286抑制GSK-3β后可部分逆转神经突的损伤,尽管轴突长度未完全恢复至正常状态,但神经元极性结构得以维持。笔者推测这可能与在体外模型中,神经元生长的环境单一,缺乏其他细胞合成的促修复和发育的蛋白分子,这在一定程度上影响了损伤的修复。
总之,体外培养神经元OGD后PI3K/Akt的活性发生变化,通过调控下游蛋白GSK-3β的磷酸化参与神经突损伤调节机制,干预PI3K/Akt/GSK-3β信号通路可能有助于受损神经突的修复。
