JAK2突变与骨髓增生异常综合征的关系
2018-10-17周旭艳牧启田欧阳桂芳
周旭艳 牧启田 欧阳桂芳
Janus激酶(JAKs)是一类非受体型可溶性的胞质蛋白酪氨酸激酶,共有4个成员:JAK1、JAK2、JAK3和TyK2,大约介导60种细胞因子和激素信号,细胞外信号与激酶的相互作用决定了JAKs的生物学功能[1]。JAK2是JAKs中与血液疾病关系最为密切的激酶,JAK2基因突变可影响骨髓造血细胞的信号传导通路,对血液疾病的发生、进展意义重大[2]。目前JAK2-V617F突变被认识到与骨髓增殖性疾病(MPDs)关系最密切[3],然而多项研究发现JAK2突变在骨髓增生异常综合征(MDS)的恶性发病机制中也起到重要作用,伴有JAK2突变的MDS患者往往预后不良[4-8]。本文就JAK2、JAK2/STAT信号通路及JAK2与MDS的关系等综述如下。
1 JAKs的结构和活性改变
JAKs由7个不同长度的JAK同源区(JAK homology,JH)(JH1-JH7)构成,包含 4 个功能区域,分别是 N-端的FERM区、SH2区、假酪氨酸激酶结构域(pseudokinase,PK)和C-端催化活性氨基酸激酶结构域(tyrosine kinase,TK)。FERM区由JH5-JH7和小部分JH4组成,调节催化活性,此区域的突变会改变JAKs蛋白激酶活性。SH2区包括大部分JH4和小部分JH3,对JAKs的活化起辅助作用。FERM区和SH2区共同作用激酶与其同源受体的相互结合,影响胞内信号传导。非功能性PK区(JH2)负性调节JAKs蛋白激酶活性,JH2功能的缺失影响细胞因子诱导的通路信号,造成机体严重的联合免疫缺陷。TK区位于JH1,具有催化活性,此区域位点的突变并不多见。最常见的JAK2-V617F突变发生于JH2同源区,JAK2-V617F突变时JH2结构区构象随之发生改变,对JH1的抑制作用减弱,使JH1磷酸化而被持续激活,从而异常启动下游信号通路[1,9],见图1。
JAKs的活性受到JAKs自身磷酸化位点和与之作用的蛋白调节。自身磷酸化位点包括负性调控位点和正性调控位点,其中负性调控位点包括Ser523、Tyr317、Tyr570 等,正性调控位点包括 Tyr1007、Tyr1008、Tyr221、Tyr813等。影响JAKs活性的蛋白包括SHP1、SHP2、PTP1B、TCPTP、CD45等蛋白质酪氨酸磷酸酶,它们通过使JAKs位点发生脱磷酸化,而影响后者活性。此外SOCS1蛋白、SOCS3蛋白和SH2B家族蛋白质对JAKs有直接的负性调控作用[1],见图1。

图1 JAKs结构模式图
2 JAK2基因突变类型
JAK2基因长 140kb,位于染色体 9p24,编码由1 132个氨基酸组成的信号转导蛋白,JAK2的突变会影响骨髓造血细胞的信号传导通路。JAK2基因点突变中以JAK2-V617F(JAK2基因第14号外显子编码序列的第1 849位碱基G被T取代,导致JH2假激酶区617位置缬氨酸转变为苯丙氨酸)突变率最高,多见于真性红细胞增多症(polycythemia vera,PV)[3]。JAK2-V617F等位基因负荷超过50%是PV患者病情进展、合并骨髓纤维化(myelofibrosis,MF)的高危因素[10]。而 MDS 患者的JAK2突变也以JAK2-V617F最为常见。其他类型点突变如JAK2-R683S(G)多见于急性B淋巴细胞白血病[11-12],总体发生率低。JAK2的12号外显子碱基的插入和缺失突变多见于PV,一般发生在第537至543位密码子之间,其中以N542-E543del的发生率最高(30%)。相比于JAK2-V617F突变,12号外显子异常的患者血象中Hb水平更高、PLT和WBC数更低,但血栓形成、继发骨髓纤维化和病情进展的概率相似[13]。JAK2基因重排的类型包括TEL/ETV6-JAK2、PCM1-JAK2、BCR-JAK2等,在MDS患者中的发生频率低。JAK2基因扩增在血液恶性肿瘤中非常少见,在MDS患者中未见报道。
3 JAK2与信号传导及转录激活因子途径(STAT)
人类STAT家族共有6个成员:STAT1、STAT2、STAT3、STAT4、STAT5(STAT5a、STAT5b)、STAT6,以 STAT3和STAT5蛋白与淋巴和骨髓恶性肿瘤的发病机制最为相关。STAT5包含STAT5a和STAT5b两种蛋白,主要在造血干、祖细胞和成熟细胞群的信号转导中发挥作用[14]。破坏STAT3和STAT5之间的生理平衡也可导致血液恶性肿瘤的发生[15]。
JAK/STAT途径本质上是一个超过30种跨膜蛋白的超家族。作为细胞因子受体信号转导的中心,JAK2/STAT可识别特定的细胞外信号因子,向胞内传递增殖、分化、抗凋亡等信号,对造血系统、免疫反应的调节至关重要[16]。JAK2的突变状态可影响信号传导。当JAK2为野生型时,只有与细胞外配体结合,才能激活细胞内STAT、PI3K和RAS-MAPK等信号通路,进而调节基因转录和细胞的增殖、分化;当JAK2发生突变时,即使没有细胞外任何配体,也可持续性激活STAT等多条信号通路[16],见图2。除JAK2自身发生的突变外,负性调节因子的突变或沉默、JAK2致癌融合蛋白的存在及调控因子含量的改变等均可激活STAT途径。国外一项研究用小鼠模型观测了JAK/STAT信号通路与负性调控因子CD45和靶分子转移蛋白受体CD71之间的关系,结果发现低表达的CD45和CD71致JAK/STAT通路持续性激活,最终导致MDS造血异常的发生[17]。
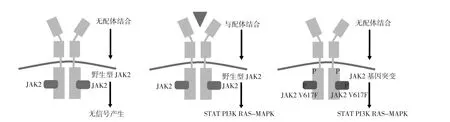
图2 野生型JAK2和突变型JAK2信号通路激活模式图
4 JAK2突变与MDS的关系
4.1 JAK2的突变率 一般认为JAK2基因以JAK2-V617F突变的形式发生于MPDs最为常见,其中在PV患者中的突变率约为95%,原发性血小板增多症和原发性MF患者中的突变率分别约为50%,而在MDS和急性髓细胞白血病患者中的突变率<5%[18-19]。
陈婉等[4]报道在中国人群中,MDS总体的突变阳性率为5%,难治性血细胞减少伴多系发育异常、难治性贫血伴原始细胞增多和不能分类的MDS亚型均检出JAK2-V617F突变,各亚型之间频率并无差异。Jekarl等[20]在检测非骨髓增殖性肿瘤的血液疾病患者基因突变时发现,MDS中JAK2-V617F突变率为8.3%。这些结果提示,尽管在MDS中JAK2突变率并不是很高,但与MDS的发病机制有一定相关性。
4.2 JAK2突变与MDS病情进展及预后 JAK2突变在MDS病情进展中起到一定作用。伴JAK2-V617F突变的MDS患者骨髓原始细胞比例、外周血乳酸脱氢酶水平高,脾脏肿大发生率高,并且约有37.5%的JAK2突变患者会发生急性白血病转化,远高于野生型患者(5.9%)[4]。此外,JAK2突变可以在MDS转白期间获得,并且可能在低危MDS亚型的病情进展中起作用[5]。国内一项临床研究对比了MDS患者和健康志愿者外周血JAK2-V617F基因的表达水平,结果发现JAK2-V617F基因表达拷贝数在恶性程度高的MDS类型中明显升高,提示MDS的发病可能受JAK2信号通路异常的影响[6]。综合上述结果,JAK2-V617F突变水平检测可作为MDS病情评价的一个指标。
JAK2突变与MDS合并MF也有密切关系。研究发现,在MDS合并MF患者中JAK2-V617F突变的发生率(21%)远大于无MF者(2%)。临床中合并MF的患者更易发生肝脾肿大,更倾向于出现消耗性症状和输血依赖,其生存率明显偏低,并且病情进展速度快[7]。Fu等[7]发现MDS合并骨髓纤维化患者JAK2-V617F等位基因负荷的中位值为22.5%(5.5%~46%),低于原发性骨髓纤维化(47%)。因此,检测JAK2-V617F的基因负荷有助于区别原发和继发性骨髓纤维化,基因负荷高的骨髓纤维化患者更倾向于原发性骨髓纤维化,若在MDS患者中检测到高负荷的JAK2-V617F突变需警惕合并骨髓纤维化可能,提示预后不良。尽管MDS合并骨髓纤维化的JAK2-V617F等位基因负荷明显低于原发性骨髓纤维化,但JAK2-V617F突变的存在有可能加速MDS患者骨髓纤维化的进程,同时影响骨髓移植的治疗效果。临床资料显示JAK2突变也是MDS患者移植后存活率低的预测因子之一,当MDS患者伴有JAK2突变时,移植后生存期明显低于无JAK2突变者,在需要骨髓移植的MDS患者中检测JAK2突变对评估预后有益[8]。
4.3 JAK2突变与MDS的治疗 目前MDS治疗方案主要包括支持治疗、低甲基化剂(阿扎胞苷、地西他滨等)、生物反应调节剂和免疫抑制剂(抗胸腺细胞球蛋白、环孢素和来那度胺等)、化疗及异基因造血干细胞移植等[21-24],但上述治疗方案在临床上仍存在诸多问题,因此,针对JAK2及其信号通路的靶向治疗是MDS潜在治疗手段之一。
卢索替尼是一种可口服的选择性JAK1/JAK2激酶抑制剂,是首个被美国FDA批准用于治疗中高危MF患者的药物。在多组临床研究中证实卢索替尼可明显缩小原发性MF患者的脾脏体积,改善患者的临床症状和生活质量[25-26]。在PV患者中,卢索替尼有明显控制血细胞比容、减小脾脏体积、减少血栓栓塞事件发生的作用[27]。此外,有相关报道称,伴有JAK2基因融合的骨髓增殖性肿瘤、慢性白血病以及骨髓增生异常综合征/骨髓增殖性肿瘤的患者在使用卢索替尼后,肿瘤负荷获得明显的改善[28-29]。但目前临床上对卢索替尼治疗伴有JAK2基因突变或合并MP的MDS患者的疗效尚不明确。Junya等[30]发现JAK2选择性抑制剂NS-018可优先抑制MDS-BMMNC(骨髓单核细胞)的增殖,有效预防MDS患者恶性克隆的过度增殖,并且对正常BMMNC产生的毒性较低。因此,NS-018有望被运用于临床治疗具有增殖性表型高危的MDS患者。
Boehrer等[31]通过体外细胞实验发现表皮生长因子受体抑制剂厄洛替尼具有抗肿瘤活性。厄洛替尼能够通过脱靶抑制作用破坏JAK2/STAT5通路的信号传导,杀死来自MDS和急性髓细胞白血病患者的CD34+骨髓原始细胞,同时能够保留正常的CD34+祖细胞。
研究发现,通过单药地西他滨化疗可降低MDS患者JAK2-V617F突变的表达水平,并且通过增加化疗周期可使拷贝数进一步降低[32]。JAK2突变的高表达可能导致MDS发生从低型向高危型恶性转化,去甲基化药物对此机制有抑制作用,而JAK2-V617F拷贝数的变化或许可以预示去甲基化药物的治疗效果。
西达本胺是一种新型苯甲酰胺类的组蛋白去乙酰化酶抑制剂,具有抗肿瘤活性[33]。Zhao等[34]在细胞实验中证实了这种抗肿瘤活性也作用于MDS,西达本胺可上调SOCS3的表达水平从而抑制MDS细胞JAK2/STAT3信号通路的传导,并且可能通过JAK2/STAT3传导信号的下调诱导MDS细胞G0/G1期停滞及凋亡。因此,西达本胺除了用于治疗难治复发的外周T细胞淋巴瘤外,还有治疗MDS的可能性,这一观点也得到其他研究者的支持[35]。
5 小结与展望
JAK2突变与MPDs的关系已经广为临床认可。JAK2-V617F突变在MDS的恶性发病机制中起到重要作用,且此类MDS患者具有独特的临床表现,往往预后不良。JAK2突变及其信号通路异常与MDS的关系是目前临床关注的热点。
安全有效的JAK2及其信号通路抑制剂正在不断地被研发,并取得了较好的临床效果。尽管如此,JAK2抑制剂治疗伴JAK2基因突变MDS患者的分子机制和临床有效性研究仍然少见。目前已发现多种治疗MDS药物的起效机制可能与JAK2/STAT途径的改变有关,但此类研究多在细胞实验中进行,临床试验少见。因此,真正实现把JAK2及其信号通路作为靶点来治疗MDS这一目标,仍任重而道远。
